|

|

|
|
UFR
sciences et technologie
IUP
SIAL
Biotechnologie
et bio-industries
61
Avenue du Général de Gaulle
94000
Créteil
Tuteur
de stage: M. Philippe Urban
|
Département
Structure et dynamique des génomes
Unité
génomique des microorganismes pathogènes
25-28
rue du Dr Roux
75015
Paris
Maître
de stage : Mme Carmen Buchrieser et M. Philippe Glaser
|
Mise au point d’une puce à ADN pour le
typage et l’étude de la biodiversité de Listéria
Julien
Tap Rapport de stage Mars Août 2005
Je
tiens tous d’abord à remercier Franck Kunst, responsable de la génopole de
l’institut Pasteur, de m’avoir accueilli et fait confiance pour ce projet
d’étude.
Je
présente également mes vifs remerciements à Carmen Buchrieser, responsable de
projet sur Legionella, et Philippe Glaser, co-responsable de l’unité
Génomique des Micro-organismes Pathogènes, pour m’avoir guidé en rendant mon
stage très enrichissant et pour m’avoir transmis de précieuses connaissances
pour l’orientation et l’aboutissement de mon projet.
J’adresse
mes remerciements à Sandra Duperrier, Elisabeth Couvé et Christophe Rusniok,
techniciens de l’unité, de m’avoir aiguillé tout au long de mes manipulations in
silico et in vitro.
Merci
enfin à toutes les personnes du laboratoire pour leur accueil dans l’unité.
Listeria monocytogenes est une bactérie
pathogène responsable d’épidémies liées à la contamination de produits
alimentaires et mortelle dans 30% des cas. Treize sérovars ont été décrit mais
les souches de sérovars 4b restent majoritaires dans les cas de listériose et
paradoxalement ce sont les souches du sérovars 1/2a qui sont le plus
fréquemment isolées dans l’alimentation. Pour investiguer la diversité
génétique des Listeria, une puce à
ADN de biodiversité pour le typage a été conçue dans l’unité Génomique des
Microorganismes Pathogènes suite aux séquençages complets des génomes et
l’analyse génomique comparative de trois souches de Listeria : L. monocytogenes
EGDe de sérovars 1/2a, L. innocua 6a
et de L. monocytogenes de sérovars 4b
Basé
sur le principe de l’hybridation moléculaire des brins d’ADN par
complémentarité, la puce d’hybridation ADN/ADN est constituée d’amplicons de
gènes déposés, appelés sondes, sur une membrane de nylon. L’obtention d’un
signal pour chaque sonde subit une binarisation afin de prédire leur présence
ou leur absence dans l’ADN extrait d’un isolat de Listeria.
Avec
l’hybridation d’ADN de plusieurs centaines de souches, la constitution d’une
base de données a permis par l’intermédiaire du regroupement hiérarchique de
confirmer les divisions génomiques observées par l’électrophorèse en champ
pulsé. Cependant, la confrontation d’un signal continu à la binarisation
entraîne la formation de valeurs ambiguës proches du seuil de coupure. De plus,
en se regroupant avec les souches de sérovars 1/2c, EGDe n’est pas
représentatif du sérovars 1/2a.
En
mettant à profit sur deux générations de puces de typage une nouvelle puce a
été réalisée en reduisant le nombre de sondes responsable de valeurs ambiguës
et en diversifiant son contenu génétique afin d’être plus représentatif de la
diversité de l’espèce. Une sélection de sondes donnant les signaux les plus
tranchés et l’intégration des gènes spécifiques à deux génomes de Listeria séquencés par TIGR (The
Institut of Genomic Research) ont été effectuées.
Réduite
et diversifiée, cette nouvelle puce, aussi discriminante que les puces
précédentes, permet de séparer les souches épidémiques très proches, en partie
grâce à un apport plus important de gènes de phage.
Mots
clés : Listeria, puce à ADN, binarisation/valeurs ambiguës, diversifier et
discriminer.
Listeria monocytogenes is a foodborne pathogen with a mortality rate of about 30%.
Intriguingly, among the 12 serovars identified within the species L. monocytogenes the majority of human listeriosis, as well as
numerous sporadic cases, have mainly been caused by L. monocytogenes serovar 4b, although strains of serotype 1/2a are most frequently isolated from foods. To
investigate the genetic diversity of the genus Listeria, a DNA array was designedUnit of Génomics of Microbial
Pathogens, based on the complete genome sequence and a comparative genomic
analysis of three Listeria
strains : L. monocytogenes
EGDe serotype 1/2a, L. innocua
serotype 6a and L. monocytogenes
serotype 4b
Based on the principle of DNA/DNA
hybridization based on its complementarity, the Listeria DNA array is constituted of probes, corresponding to
specific genes, spotted on a nylon membrane. In order to predict the presence
or absence of a gene from the DNA extracted from the investigated Listeria isolate, the signal obtained
for each probe is translated into binary scores.
The hybridization results for the
DNA of several hundreds of Listeria
strains was collected in a data base which allowed by using hierarchical
clustering to confirm the genomic divisions identified earlier by pulsed field
electrophoresis. However, the translation of the hybridization signals into
binary scores leads also to the generation of ambiguous values close to the
threshold. Moreover, the used reference strain EGDe is not representative of
serotype 1/2a. strains, but clusters with serotype 1/2c strains.
Based on the results, obtained from
hybridizations of two previously designed Listeria
DNA arrays, a third Listeria array
was developed, by reducing the number of probes generating ambiguous values and
by diversifying the genetic information in order to be more representative of
the diversity present within the species. A selection of probes giving the most
distinct signals and the integration of specific genes of two additional Listeria genomes sequenced by TIGR (The
Institute of Genomic Research) was carried out.
This new reduced and diversified Listeria DNA array, is as discriminating
as the preceding ones, allows to separate very closely related epidemic
strains, partially due to the selection of more phage-related genes.
Key words: Listeria, DNA array, binary scores/ ambiguous values,
diversification and discrimination.
Sommaire
Remerciements
Résumé
Abstract
Sommaire
I. Introduction - Revue bibliographique.............................. 1
A. Présentation de Listeria
monocytogenes............................................................ 1
1. Caractéristiques et physiologie de Listeria monocytogenes.................................................................. 1
2. Taxonomie................................................................................................................................................................ 2
3. Du point de vue génomique................................................................................................................................. 2
4. Epidémiologie.......................................................................................................................................................... 4
B. Méthodes de typage............................................................................................................................ 5
1. Sérotypage............................................................................................................................................................... 6
2. Lysotypage................................................................................................................................................................ 6
3. Ribotypie................................................................................................................................................................... 7
4. Électrophorèse en champs pulsé (PFGE)....................................................................................................... 7
5. Le typage par séquençage de sites multiples (MLST).............................................................................. 8
C. Les puces à ADN pour le typage de L.
monocytogenes.................................. 8
1. Contexte du développement de ces puces.................................................................................................... 8
2. Principe des puces à ADN................................................................................................................................. 11
3. Analyse génomique : présence ou absence de gène................................................................................ 12
4. Conception de la puce biodiversité Listéria.............................................................................................. 13
D. Puce biodiversité de 3ème
génération......................................................................... 16
1. Analyse et utilisation d’une puce à ADN pour le typage
génomique................................................. 16
2. Comparaison et classification des souches............................................................................................... 20
3. Difficultés et solutions envisagées.............................................................................................................. 22
4. La puce de typage de 3eme génération...................................................................................................... 23
II. Matériels et méthodes................................................. 25
A. Souches bactériennes.................................................................................................................. 25
1. Souches matrices................................................................................................................................................ 25
2. Souches testées sur la nouvelle puce.......................................................................................................... 25
B. Données utilisées pour la sélection
des gènes.......................................... 26
C. Construction de la puce............................................................................................................ 27
1. Dessin des oligonucléotides............................................................................................................................ 27
2. PCR en masse des gènes sélectionnés.......................................................................................................... 27
3. Vérification des produits de PCR par séparation par
électrophorèse sur gel d’agarose........ 28
4. Dépôt des produits PCR sur membrane de nylon..................................................................................... 28
D. Utilisation de la puce................................................................................................................. 30
1. Marquage radioactif de l’échantillon d’ADNg......................................................................................... 30
2. Hybridation........................................................................................................................................................... 32
3. Analyse des membranes.................................................................................................................................... 34
III. Résultats et interprétations 35
A. Sélection des gènes à déposer........................................................................................... 35
1. Simulation « in silico »..................................................................................................................................... 35
2. Stratégie de la sélection des gènes............................................................................................................. 39
3. Représentativité des gènes.............................................................................................................................. 41
B. Caractéristique et influence du
matériel........................................................ 42
1. Étude de concentration minimale et nécessaire pour la
PCR.............................................................. 42
2. Comparaison des signaux d’hybridation en fonction de
la qualité de produits PCR spot....... 43
3. Influence de la température sur l’hybridation........................................................................................ 44
4. Influence des différents lots de membrane............................................................................................ 46
C. Utilisation de la puce 3G......................................................................................................... 48
1. Comparaison de kits d’extractions d’ADNg à analyser......................................................................... 48
2. Séparation des sérovars et division génomique....................................................................................... 49
3. Sous groupage de souches d’un même sérovars....................................................................................... 50
IV. Discussions.............................................................. 52
A. Critique de la stratégie de
sélection de gènes........................................... 52
B. Influence du matériel.................................................................................................................. 52
C. Fiabilité de la puce 3G................................................................................................................... 53
D. Comparaison avec l’ancienne membrane................................................................ 53
Conclusion.................................................................... 54
Bibliographie................................................................. 55
Tables des illustrations.................................................. 58
Le potentiel pathogène de Listéria et en particulier
de l’espèce L. monocytogenes est dû à ses caractéristiques
physiologiques et génomiques. Listeria à l’origine de la listériose,
maladie mortelle pour 30% des cas, pose un réel problème de santé publique et
pour les industries agro-alimentaires [1]. Les différents programmes de recherche où s’inscrit
l’unité de Génomique des Microorganismes Pathogènes ont pour objectif de mieux
comprendre cette bactérie et la maladie qu’elle provoque et d’améliorer la
prévention des contaminations notamment par le développement des puces de
typage et d’analyse génomique « biodiversité Listéria ».
Listeria a été décrite pour la première fois dans les
années 20, mais ce n'est que depuis la mise en évidence d'une origine alimentaire
de la listériose humaine, lors d'une épidémie survenue au Canada en 1983, que
cette maladie est reconnue comme un véritable problème de santé publique [5]. Cela a conduit un certain nombre de pays à instaurer
des systèmes de surveillance de l'infection, basée essentiellement sur la
centralisation et la caractérisation des souches cliniques de L. monocytogenes
dans un laboratoire [en France, il s'agit du Centre National de Référence des Listeria
(CNR Listeria) localisé à l‘Institut Pasteur de Paris]. En France, la
listériose est depuis mars 1998 une maladie à déclaration obligatoire [1].
Listeria monocytogenes est un bacille gram + à faible
GC%, non sporulante, d’environ 0,4 μm de diamètre et de 0,5-2 µm de
longueur, elle peut être présente sous forme isolée ou en petites chaînettes. [1]
Ce germe pathogène est particulièrement résistant à des
conditions difficiles de croissance. En effet, il s’agit d’un bacille
psychrotrophe, il peut se développer à des températures de 1 à 45°C, ce qui lui
permet de se multiplier dans les chambres froides. Il résiste aussi à la
congélation. Cette faculté d’adaptation au froid est une caractéristique qui
contribue à sa survie et sa multiplication dans les aliments, car il constitue
un avantage par rapport aux autres bactéries lors de la conservation au froid.
Il faut tout de même noter que le temps de génération de L. monocytogenes
est plus faible à des températures de 4 à 8°C qu’à 30- 37°C. L. monocytogenes
est uniquement mobile à
20-25°C. [1]
En plus de son adaptation au froid, la L. monocytogenes
peut se développer dans des milieux riches en sels (elle est dite halophile),
elle peut se multiplier dans des milieux pauvres en eau, et en oxygène
puisqu’il s’agit d’une bactérie anaérobie facultative. Sa croissance est
possible à des pH oscillants entre 6 et 9. Ces différents paramètres
correspondent aussi aux caractéristiques des aliments sensibles à Listeria
comme les fromages et la charcuterie. L'acidification du milieu permet de
partiellement inactiver le germe, et de stresser les cellules survivantes, de
sorte que la reprise de croissance est retardée.[1]
Cependant, même si ces bactéries paraissent très
résistantes, une pasteurisation standard (30 min à 62°C) permet de les
éliminer. Elles ne survivent pas à la plupart des désinfectants (les
aldéhydes, les dérivés chlorés…).[1]
Le genre Listeria comprend deux espèces pathogènes, L.
monocytogenes et L. ivanovii (pathogène des ruminants), et quatre
non pathogènes : L. innocua, L. seeligeri, L. welshimeri
et L. grayi. Il existe 13 sérovars pour Listeria monocytogenes,
mais ce sont majoritairement les types 4b, 1/2a et 1/2b qui provoquent la
listériose (voir Tableau
1). Les souches du sérovars 4b semblent avoir un
potentiel pathogène plus important car malgré le fait qu’elles sont rarement
isolées dans la nourriture, on les trouve fréquemment responsable de cas de
listérioses.[1]
Les chromosomes de listeria analysés à ce jour sont
circulaires et d’une taille d’environ 3 000 000 paires de base avec
un taux de G+C% de 37 à 39%. La souche de L. innocua qui a été séquencée
contient aussi un plasmide de 81kbp. 2853 gènes codant pour des protéines ont
été prédits dans le génome de la souche EGDe de L. monocytogenes. [12]
Le génome de Listeria code pour un grand nombre de
protéines des familles suivantes : des protéines de surface à motif LPXTG
(voir Figure 1), des internalines, des systèmes de transport de
sucres, des protéines similaires à des protéines de compétence de Bacillus
subtilis et des régulateurs de la famille Crp/Fnr. La comparaison des séquences
génomiques de L. monocytogenes et L. innocua est a été
et est toujours utilisé pour identifier
des gènes impliqués dans la virulence de L. monocytogenes et, plus
généralement, à mieux comprendre la pathogénicité de L. monocytogenes
et sa capacité à contaminer la nourriture [12]. L. innocua est aussi retrouvé dans des
aliments.
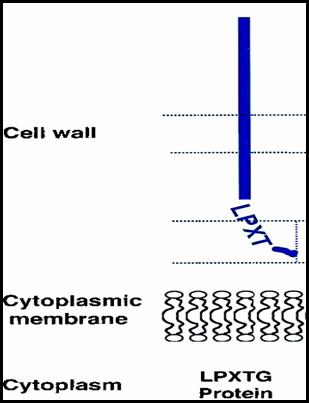
Figure 1 : Protéine de surface liés par covalence
avec la paroi cellulaire contenant un motif LPxTG
Les deux génomes codent pour de nombreuses protéines de
surface. Pour L. monocytogenes, 41 protéines contiennent un motif
C-terminal Leu- Pro- X- Thr –Gly (LPXTG) qui permet d’avoir
une liaison covalente avec le peptidoglycane (Figure 1). C’est à ce jour, la
bactérie qui code pour le plus de protéines de cette famille. Parmi ces
protéines, on retrouve les protéines de la famille des internalines (Inl). Les
internalines InlA et InlB sont les mieux caractérisées. InlB présente un
domaine de liaison non covalente à la paroi. Ces deux internalines sont
notamment responsables du potentiel infectieux de L. monocytogenes en
ayant un rôle essentiel dans l’entrée dans les cellules de l’hôte.[8][12]
Le grand nombre de 331 gènes codant pour des transporteurs
protéiques sont probablement impliqués dans la capacité de la colonisation et
la croissance de Listeria dans un large panel d’écosystème ainsi que les
protéines du système PTS (Système Phosphotranferase-Phosphoenolpyruvate dépendant)
qui doivent permettre à Listeria d’utiliser une grande diversité de
sucres. La proportion élevée de gènes régulateurs (Crp/Fnr ; GntR ;
BglG) à hauteur de 7,3% peut expliquer la capacité d’adaptation à des
environnements très divers de L. monocytogenes. [12]
Quatre familles de protéines de stress ont pu êtres
identifiés, leurs fonctions ont pu être établi dans les conditions de choc aux
températures froides, mais également pour la résistance à l’acidification du
milieu. Le génome de L. monocytogenes contient également des gènes
de dégradation des sels biliaires. De manière intéressante, ces gènes sont
absents chez L. innocua. [12]
Listéria est à l’origine de la listériose. Infection
essentiellement d'origine alimentaire, la listériose est diagnostiquée
principalement dans les pays industrialisés. L. monocytogenes est
un pathogène opportuniste. Les personnes à risques sont les sujets dont le
système immunitaire est immature ou perturbé, c'est à dire, les femmes
enceintes, les nouveau-nés, les personnes âgées et les patients immunodéprimés
suite à une maladie ou à un traitement immunosuppresseur. [1]
Cette infection se traduit par des formes invasives :
bactériémie/septicémie et infection du système nerveux central chez le
nouveau-né et l'adulte, et des avortements chez la femme enceinte. Le
renforcement des procédures de surveillance et d’informations ont permis de
diminuer l’incidence à moins de 4 cas par million d'habitants en France en l’an
2000. Néanmoins, la maladie est caractérisée par une mortalité élevée[21]. Plus récemment, des gastro-entérites à Listeria
monocytogenes ont été décrites, mais, cette forme de listériose non
invasive n'a pas été décrite en France à ce jour.[1]
En France, d'après le plan de surveillance de la Direction
Générale de la Concurrence, de la Consommation et de la Répression des Fraudes
réalisé au stade de la distribution, les aliments les plus fréquemment
contaminés par Listeria monocytogenes sont les charcuteries
cuites (langues, têtes, rillettes…), les produits de saucisserie, les graines
germées réfrigérées, et certains produits laitiers (fromages à pâte molle et au
lait crû).
À l'Institut Pasteur, le Centre National de Référence des Listeria
est chargé de la surveillance microbiologique de la listériose humaine. Lors
d'épidémies, il participe au dispositif associant la Direction Générale de la
Santé, l'Institut de Veille Sanitaire, la Direction Générale de l'Alimentation
et la Direction Générale de la Concurrence, de la Consommation et de la
Répression des Fraudes.
La listériose évolue sous forme de cas sporadiques, auxquels
peuvent s'ajouter des cas groupés voire des épidémies. Des cas de listériose
sont dits groupés si au moins deux cas, dus à des souches non distinguables par
une analyse en champs pulsé, surviennent dans un intervalle de temps inférieur
à 6 mois, chez des personnes ayant fréquenté un même lieu d’exposition. Si
l'intervalle de temps entre les cas est supérieur à 6 mois, on parlera de cas
liés qui ont une importance épidémiologique moindre que les cas groupés. En
France, en 2001, le CNR a recensé les souches de 185 cas sporadiques de
listériose, soit le nombre le plus faible depuis 1987[21]. Ce nombre de cas est relativement stable depuis
1996. Avant cette date, le CNR a identifié jusqu'à environ 750 cas en 1992,
dont 279 cas épidémiques liés à la consommation de langue de porc en gelée[17]. Le nombre de cas sporadiques a donc fortement
diminué depuis 10 ans. Ceci résulte à une meilleure surveillance par les
industriels et un control administratif en particulier.[1]
Du fait de la présence fréquente de L. monocytogenes dans
l'environnement, il est très difficile d'empêcher toute contamination au niveau
de la production des aliments. Afin d'améliorer la gestion de ces
contaminations, les industriels sont intéressés par des méthodes de détection
et de typage plus performantes et plus rapides [1][14].
La caractérisation des souches de Listéria est très
importante pour les investigations épidémiologiques. Elles permettent de mettre
en évidence rapidement les cas groupés et les épidémies. Par ailleurs, elles
sont aussi utilisées pour identifier l’origine alimentaire de cas de
listériose. Par conséquent, de nombreuses méthodes de typages ont été décrites
pour ce micro-organisme.
Le typage de Listéria monocytogenes a évolué en même
temps que les progrès des techniques de biologie moléculaire. De la technique
du sérotypage, la plus ancienne, suivirent la technique de lysotypage, le
ribotypage puis l’électrophorèse en champs pulsé. Le but recherché est d’être
de plus en plus discriminant en mettant à profit la biodiversité des Listeria.
Ces méthodes permettent de classer les isolats et de définir des relations
entre ces isolats. En particulier elles doivent permettre de dire si deux
isolats ont la même origine et d'aider à l'identification de la source d'une
épidémie ou de cas sporadiques.
Les souches sont sérotypées
en routine. C’est une technique facile à mettre en œuvre reposant sur le
principe d’agglutination des bactéries sur sérums. Les combinaisons des
facteurs antigéniques représentés dans le Tableau
1 définissent le sérotype. Deux types d’antigènes sont
reconnus par ces sérums : les antigènes flagellaires et les antigènes
somatiques (sans doute les acides teichoïques).
A ce jour, les déterminants génétiques de ces sérotypes sont
encore largement inconnus. Les cas cliniques ont souvent été associés aux L. monocytogenes
des sérotypes 4b, 1/2a et 1/2b. Cependant au-delà de l’aspect taxonomique de la
technique, le sérotypage a une valeur épidémiologique limitée au vu de la
biodiversité des listéria au sein d’un même sérovars. C’est donc dans ce but
que d’autres techniques plus discriminantes ont été développées.
Tableau 1:
Sérotypes au sein du genre Listeria
|
Espèce
|
Désignation
|
Antigène O (somatique)
|
Antigène H (flagellaire)
|
|
L. monocytogenes
|
1/2a
|
I ; II ; III
|
A ; B
|
|
1/2b
|
A ; B ;C
|
|
1/2c
|
B ; D
|
|
3a
|
II ;III ;IV
|
A, B
|
|
3b
|
A, B, C
|
|
3c
|
II ; III ;IV ; XII ; XIII
|
B, D
|
|
4a
|
III ; V ;
VII ;IX
|
A ; B ; C
|
|
4b
|
III, V, VII, IX
|
|
4c
|
III ; V ; VII
|
|
4d
|
III ; V ; VII
|
|
4e
|
III ; V ; VI ; VIII ;IX
|
|
7
|
III ; XII, XIII
|
|
L. ivanovii
|
5
|
III ; V ; VI ; VIII ; X
|
|
L. innocua
|
6a
|
III ; V ; VI ;
VII ;IX ;X ;XI
|
|
6b
|
III ; XIV
|
|
L. grayi
|
|
|
E
|
La lysotypie est une technique qui n’est plus utilisée pour
caractériser le genre Listeria du fait de problèmes d’interprétation, de
reproductibilité et de discrimination. Le principe de la lysotypie repose sur
la sensibilité aux phages. Une bactérie peut être résistante aux phages soit
parce qu’elle est déjà lysogène soit parce qu’elle n’a pas de récepteur pour ce
phage. Ceci est dû à la présence de différents types d’acide téichoïque entre
les différents sérovars qui influent sur la sensibilité aux bactériophages. Par
exemple, les phages A118 et PSA sont respectivement et spécifiquement
infectieux pour les sérovars du groupe 1/2 et 4[19][29].
La ribotypie est couramment utilisée. Se basant sur la
restriction de l’ADN total et l’hybridation à l’aide d’une sonde correspondant
aux sous unités ribosomales 16s ou 23S, la ribotypie permet de mettre en
évidence le polymorphisme des souches [11]. Cette technique peut être facilement standardisée
grâce à son automatisation par le riboprinter® [27]. Cependant, le pouvoir discriminatoire de la technique
est faible, notamment au niveau des souches de sérotype 4b. De plus, la
ribotypie ne donne aucune information génomique ou fonctionnelle sur la souche
et son coût est élevé.
A ce jour la PFGE est
la technique la plus discriminante pour typer les souches de L. monocytogenes
et est utilisée en routine dans la plupart des laboratoires dans le monde dont
le CNR Listéria à l’Institut Pasteur. Elle permet, au niveau des industries
agroalimentaires, une bonne traçabilité des souches environnantes, en
connaissant leur pulsotype unique, et évaluer la maîtrise de qualité hygiénique
lors de la désinfection des locaux. Une standardisation très poussée de la
méthode a été nécessaire pour l’échange des données entre laboratoire. Le CDC
(Control Disease Center) d’Atlanta a construit une banque de données
« PulseNet » qui regroupe tous les profils de restriction obtenu par
PFGE[18]. Chaque profil obtenu est répertorié par
l’intermédiaire d’un code et chaque nouveau profil est comparé à cette banque
en constante évolution. Celle-ci permet alors de tracer rapidement l’évolution
des clones déjà isolés et de mettre en évidence l’émergence de nouvelle lignée.
[10]
Cependant la PFGE peut apparaître comme trop discriminatoire
et biaiser les conclusions en différenciant une souche clinique d’une souche
environnementale qui peuvent être séparées a posteriori par la présence ou non
d’un prophage intégré. De plus, les profils de restriction obtenus ne donnent
pas de renseignement sur les gènes présents si cette technique n’est pas
complétée par une analyse par Southern blot.
Le MLST (Multilocus sequence typing) repose sur le
séquençage de plusieurs gènes, en général sept, conservés au sein d’une espèce.
L’avantage de cette technique est la possibilité de comparer les résultats
entre différents laboratoires et de donner des relations phylogénétiques entre
les souches. Dans le cas de listeria, cette méthode est moins
discriminante que l'analyse en champs pulsé
[26]. Par ailleurs le MLST est assez cher et lourd à
mettre en place et ne peut être réalisé que dans des laboratoires équipés pour
le séquençage.

Figure 2 : Représentation des différentes lignées à
partir d’un PFGE numérisé de 62 souches représentatives de L. monocytogenes
[2]
La Figure
2 montre les deux principales divisions génomiques de L. monocytogenes
identifié par la similitude des profils obtenu par PFGE. Il faut noter la
corrélation entre les sérovars et ces différentes lignées définies sur la base
de la structure du génome.
La connaissance complète des
génomes bactériens a permis le développement d'un grand nombre de méthode
d'analyse dite post-génomique. Parmi ces approches globales, les puces à ADN
permettent d'analyser en une seule étape un grand nombre d'acides nucléiques.
L'application principale concerne l'analyse des ARN messagers d'une cellule: le
transcriptome. Les puces à ADN permettent aussi de détecter la présence ou
l'absence d'un gène et donc de caractériser les différences de contenu
génétique entre deux isolats d'une même espèce. Les puces
« biodiversité » Listeria développées au laboratoire portent
des sondes correspondant à des gènes spécifiquement présents dans différents
isolats de Listeria, mais qui ne sont pas
présents dans tous les isolats séquencés à ce jour. Elles sont établies à
partir de la comparaison des séquences de plusieurs génomes.
Par une seule expérience d'hybridation, ces puces permettent
d'établir une empreinte digitale correspondant aux gènes présents ou absents
dans un clone, et constitue donc une méthode globale de typage. A la différence
des autres méthodes de typage utilisées pour Listeria, les puces à ADN
apportent une information fonctionnelle sur la nature des gènes qui
différencient deux isolats. Ces résultats peuvent être comparés aux données
phénotypiques sur les souches, notamment en relation avec leur virulence[13].
Après avoir séquencé les génomes de L. monocytogenes
EGDe et L. innocua, l'unité GMP a développé une puce de typage pour
L. monocytogenes dans le cadre de plusieurs consortiums. Les
objectifs étaient de développer un outil de typage aussi résolutif que le champ
pulsé et qui soit automatisable. Par ailleurs, dans le cadre de la comparaison
d'un grand nombre de souches d'origine clinique ou environnementale,
d'identifier les différences entre les souches et de définir s'il est possible
d'identifier des caractéristiques spécifiques aux souches responsables de
maladies chez l'homme. Ces études doivent permettre de comprendre l'évolution
au sein du genre Listeria, de caractériser les transferts génétiques et
d'identifier de nouveaux gènes de virulence. Par ailleurs à plus long terme la
possibilité de prédire par une analyse génomique le risque associé à une
contamination peut avoir une influence sur la gestion de cette contamination.[7]
Afin de réaliser ce projet l'Unité GMP s'est associée à
différents partenaires ayant des expertises complémentaires sur la biologie des
listéria, sur l'épidémiologie et sur les problématiques spécifiques aux
industries agro-alimentaires dans le cadre de deux projets de collaboration,
l’un interne à l’Institut Pasteur (le programme transversal de recherche),
l’autre financé par le ministère de l’agriculture (le programme AQS).
a) Le programme transversal de recherche
La mise au point d’une puce
de biodiversité Listeria a commencé au cours d’un programme transversal
de recherche de l’Institut Pasteur mettant en jeu la collaboration entre trois
laboratoires. Les objectifs sont :
Ø
Le développement d'une stratégie
pour l'analyse génomique d'un grand nombre de souches de Listeria en
commençant par des souches de la collection de l'Institut Pasteur, afin
d’établir une corrélation entre les données épidémiologiques et phénotypiques
déjà collectées et les données génomiques récentes (l’épidémiologie génomique).
Ø
L'identification de gènes et/ou de
régions chromosomiques, potentiellement impliqués dans la pathogénicité et la
compréhension de leurs fonctions. En particulier, la découverte, à l'aide de
l'épidémiologie génomique, de gènes importants pour l'infection chez l'homme
qui ne sont pas révélés par les modèles animaux actuellement disponibles.
Ø
Le développement d'outils
rationnels d'identification et de typage des isolats de L. monocytogenes.
Ces outils devraient permettre de distinguer des contaminations potentiellement
dangereuses de celles certainement sans risque.
b)
Le programme AQS
Un deuxième programme d’étude de Listeria s’inscrit
dans le cadre du programme Aliment – Qualité – Sécurité (AQS), financé par le
Ministère de l’Agriculture de l’Alimentation de la Pêche et des Affaires
Rurales, où l’unité de génomique des microorganismes pathogènes répond aux
problématiques d’étude de la biodiversité de Listeria par une
corrélation des techniques de biologie moléculaire et l’étude de la virulence.
Afin de mieux caractériser le risque dans un contexte de
risque sanitaire lié aux aliments, l’unité GMP coordonne le programme qui vise
en réalisant une étude d’un ensemble de souches représentatifs des divers
habitats de L. monocytogenes (souches environnementales, d'aliments, de
portage asymptomatique, des cas cliniques) de façon à préciser leur
potentialité de virulence ou à l’inverse leur non pathogénicité.
Cette étude combine alors génomique, informatique,
protéomique, transcriptomique et l’étude de la virulence pour pouvoir mieux
caractériser les gènes responsables du potentiel pathogène de L. monocytogenes
et ainsi pouvoir prédire et hiérarchiser les risques liés à des produits
alimentaires contaminés.
Le programme d’étude AQS des Listeria s’effectue en
collaboration avec les partenaires suivants :
Ø
Unité des Interactions
Bactéries-Cellules de l’Institut Pasteur (P. COSSART)
Ø
Laboratoire des Listeria
(P.MARTIN, M.DOUMITH)
Ø
Laboratoire de Microbiologie UMR
INRA Dijon (J.GUZZO).
Ø
SRV-Microbiologie INRA de Theix (M.HEBRAUD).
Ø
Laboratoire départemental de la
Côte d’Or
Ø
Industries agroalimentaires
(Arilait, Entremont, Salaison Dijonnaise, Soredab)
La collaboration de l’ensemble de ces partenaires permet
l’association de compétences multidisciplinaires pour répondre aux objectifs
fixés. C’est dans le cadre de ce projet que s’inscrit mon stage de maîtrise.
Les puces
à ADN repose sur le principe d’hybridation moléculaire entre deux acides
nucléiques simples brins. Cette hybridation moléculaire s’effectue lorsque deux
brins sont complémentaires. Les quelques centaines de séquences d’ADN
recherchées sont déposées sur un
support. Cet ensemble constitue la puce à ADN. L’ADN à analyser va s’hybrider
selon le principe d’hybridation moléculaire sur les différents acides nucléiques
que porte constitue la puce à ADN. La détection de ces hybridations, après le
lavage de l’ADN hybridé de manière non spécifique, s’effectue soit par
détection de la fluorescence soit de la radioactivité (voir Figure 3).[13]

Figure 3 : Analyse d’acides nucléiques par puce
à ADN.[13]
Ce principe peut être alors utilisé pour l’analyse de
transcriptome en hybridant les ADN complémentaires des ARNm et également pour montrer
l’absence ou la présence de gènes dans un génome par marquage de l’ADN
génomique.
Au niveau d’une espèce bactérienne, l’analyse des génomes montre une grande diversité en contenu
génétique. Une partie du génome étant partagé par tous les isolats et le reste
étant spécifique de certains isolats. Par exemple 30% du génome de la souche d'E.
coli O157:H7 sont absents de la souche commensale K12, Ces régions
spécifiques d'une espèce peuvent correspondre à des éléments mobiles, des
transposons, des prophages ou des plasmides ou des gènes indépendants de ces
éléments. Cela peut correspondre à un seul gène, à un opéron ou à des régions
plus complexes. Les puces à ADN permettent de détecter dans un génome la présence
d'un gène pour lequel une sonde est portée par la puce. Le pouvoir de
discrimination et de caractérisation génomique dépend donc des connaissances
génomiques sur une espèce. La connaissance des génomes de plusieurs isolats
représentatifs de la diversité de l'espèce permet d'améliorer la valeur de ce
typage.
L'unité GMP a développé deux générations de puce
"biodiversité" pour listéria. Le développement de ces puces demande
dans un premier temps la connaissance de la séquence des génomes de souches
d’espèces de Listeria différentes et de sérovars différents puis d’une
étude comparative génomique afin d’identifier les gènes suffisants et
nécessaires pour permettre de discriminer les souches de listéria.
a) Séquençage et comparaison génomique
La première
séquence génomique de L. monocytogenes (souche EGDe) a été publiée
en la comparant à celle de l'espèce la plus proche non pathogène L. innocua.
En excluant les gènes de prophage, 270 gènes sont spécifiques de L. monocytogenes
et 149 gènes sont spécifiques de L. innocua (CLIP11626)[3]. Les 270 gènes spécifiques de L. monocytogenes
sont regroupés en 100 îlots répartis sur le génome. L'évolution entre les deux
souches correspond donc à un très grand nombre d'événements d'insertion ou de
délétion. Ces deux espèces étant très proches phylogénétiquement, ces gènes
constituent potentiellement de bonnes sondes pour le typage en relation avec la
virulence. EGDe est une souche historique isolée de cobaye qui a été repiquée
et distribuée dans de très nombreux laboratoires. La souche EGDe de sérovar
1/2a est un isolat particulier dont le génome a été séquencé et qui est devenue
une souche de référence. [12]
Afin de
caractériser des souches d'origine humaine et de comprendre la virulence des Listeria
pathogènes, il était important de connaître la séquence génomique d’une souche
clinique[15]. L'unité GMP a ensuite déterminé la séquence
génomique d'une souche épidémique de L. monocytogenes de sérovar 4b
(CLIP80459). Cette souche a été isolée lors d’une épidémie qui a eu lieu en
France à la fin de l’année 1999. Par ailleurs, les souches de sérovar 4b et
celles de sérovar 1/2a représentent les deux sous-groupes principaux de
l'espèce (Voir Figure 2). La connaissance de ces deux génomes permet donc de
mieux couvrir la diversité de l'espèce, cette séquence génomique a été comparée
avec celle de EGDe [16]. La comparaison de ces deux génomes a mis en évidence
une diversité du contenu génétique au sein même de l'espèce L. monocytogenes.
En effet, environ 6% des gènes sont spécifiques d'une souche par rapport à
l'autre. Afin d'avoir une connaissance génomique approfondie du genre Listeria,
en collaboration avec le consortium allemand
" Pathogenomics " l’unité GMP a déterminé aussi les
séquences des génomes de L. ivanovii, L. grayi. L. seeligeri
et L. welshimeri. Finalement, aux Etats-Unis, TIGR (The Institute of Genomic Research) a
séquencé le génome de deux souches sérovar 4b et d'une souche de sérovar 1/2a.
L'ensemble de ces informations génomiques constitue une base de connaissance
très riche pour la mise au point de puces biodiversité.
b) Les premières générations de puces à ADN
Une première puce
de biodiversité de Listeria a été construite par l'Unité GMP sur la base de la
comparaison des génomes de EGDe, L. innocua et de la séquence
partielle de L. monocytogenes 4b CLIP80459. Cette puce porte 409
sondes, 262 correspondant à des gènes de la souche EGDe, 94 à la souche de L.
innocua et 53 à la souche 4b. Cette première membrane a été utilisée pour
une étude comparative génomique de 113 souches de Listeria [7]. Les résultats d’hybridations avec cette puce de
première génération ont permis de montrer qu'elle constituait un outil de
typage performant permettant l'identification de l'espèce, le sérovar et
permettant de sous grouper chaque sérovar. La présence des gènes de virulence
connus a permis de montrer que les facteurs de virulence sont présents chez
toutes les souches de L. monocytogenes testées.[7].
Une puce de 2ème
génération a ensuite été construite en tenant compte d’autres séquences
spécifiques d’une souche L. monocytogenes du sérovars 1/2b après le
séquençage partiel d'une souche de ce sérovar
et du sérovar 4b après l'achèvement de la séquence de la souche CLIP80459
et la publication d’une souche du sérovars 4b, séquencée par TIGR. Plus
complète et plus représentative de l’espèce L. monocytogenes, cette
puce a permis de confirmer les résultats obtenus avec la première puce (cf. I.C.4.c)).
En collaboration
avec le Centre National de Référence des Listeria (Institut Pasteur), la
diversité de l'espèce L. monocytogenes et la spécificité des
souches d'origine clinique a été étudiée, par hybridation de la puce de 2ème
génération portant 739 gènes spécifiques d'au moins une des souches séquencées
(EGDe, 4b, L. innocua, 1/2b) [7]. Les résultats d'hybridation obtenus pour 263 souches
de L. monocytogenes, d'origine et de caractéristiques différentes, et
pour 20 souches représentant toutes les espèces du genre Listeria
montrent que ces puces sont un outil de typage puissant permettant de
différencier les espèces du genre Listeria et de subdiviser les souches
appartenant à l'espèce L. monocytogenes.
L'analyse génomique
utilisant les puces à ADN « biodiversité Listeria », montre
qu’il existe trois grands groupes chez L. monocytogenes en
corrélation avec le sérovar : les souches de sérovars 4b, 1/2a et
1/2b :
Ø
Les souches 1/2a clustérisent en
deux grands groupes comprenant en majorité de souches d’origine
environnementale et un autre sous groupe de souches de portage.
Ø
Les Souches 4b (cette analyse
est réalisée en ajoutant les données obtenues sur les souches analysées dans le
cadre du programme AQS 23) se divisent en 4 grands groupes avec la présence de
souches des différentes origines (épidémiques, environnementales, filière
porcine et portage) dans chacun des grands groupes. Cependant les différents
types de souches (épidémiques, environnementale, filière porcine et portage)
peuvent se regrouper sous forme de sous groupe.
d) Difficultés rencontrés
Les groupements de
souches obtenues par classification hiérarchique effectué à partir des données
acquises par l’intermédiaire des puces à ADN correspondent exactement à ceux
obtenu par PFGE. Cependant, la classification hiérarchique nécessite une
binarisation du signal obtenu par les puces à ADN. Cette observation conforte
la valeur de la classification des souches après typage par puce à ADN.
Néanmoins, pour les souches proches l'existence de valeurs proches du seuil de
binarisation donc ambigu entraîne des difficultés d’analyse. Deux souches
identiques peuvent par exemple apparaître différentes par cette analyse.
L'objectif de mon stage était la réalisation
et la validation d'une puce de troisième génération avec comme cahier des
charges de réduire le nombre de sondes en éliminant les sondes qui apportaient
peu d'information et les sondes donnant des signaux ambigus et d'augmenter la
valeur de discrimination de la puce par l'ajout de nouvelles sondes sur la base
de l'ensemble des séquences disponibles. Ce projet a été réalisé en tenant
compte de l'expérience acquise sur les deux premières puces.
Afin de comprendre quels seront les enjeux du développement
d’une nouvelle puce à ADN, il faut en rappeler les principes d’analyses et
d’utilisations. C’est à partir du ciblage des difficultés qui peuvent survenir
dans l’analyse des puces à ADN que des améliorations pourront être apportées.
Par l’intermédiaire
du marquage, radioactif par exemple, lors de l’hybridation sur la puce un
signal peut être détecté et quantifier (Voir Figure
4). Celui-ci sera ensuite normalisé et comparé à une
valeur de référence pour quantifier l’ADN ayant hybridé à la sonde fixée. Afin
de prédire l’absence ou la présence de gène au sein de l’ADNg analysé, il est
nécessaire d’effectuer un traitement du signal. La définition d’un seuil de
coupure va permettre de binariser (1= présence du gène, 0= absence du gène)
l’information.
a) Quantification du signal
Dans le cas des
puces de biodiversité, les puces sont de type macro-array avec un dépôt des
sondes sur une membrane de nylon et un marquage radioactif de l’ADN. La
quantification du signal est réalisée par activation d’écran phosphore et la
lecture par le Typhoon (appareil de type phosphorimager). Le logiciel
Array-vision est utilisé pour quantifier l’intensité de chaque dépôt.
Dans le programme
Array-vision®, différents paramètres comme la distance entre 2 taches, le diamètre des taches, le nombre de
colonnes, le nombre de lignes sont définis afin que le logiciel puisse créer
une grille d’analyse qui sera placée sur l’image scanné de la membrane.
|

Figure 4 : Scan de la membrane portant 739 sondes en duplicata
hybridée par l’ADNg de L. monocytogenes marquée radioactivement
au 33P
|

Figure 5 : Quantification des signaux avec Array-vision
|
|
Ce protocole est importé à partir d’une grille établie sous
Excel comportant les noms de l’ensemble des sondes déposéesspottées sur la
membrane, pour pouvoir faire la liaison entre les taches et les noms de
gènes.
|
Une fois le
protocole terminé, la grille est placée sur l’image de la membrane.
L’utilisateur doit vérifier que la grille est correctement placée et peut
replacer certains spots manuellement si nécessaires (voir Figure 5). Le logiciel attribue à chaque tache une valeur
numérique (unité arbitraire). Ces données sont obtenues sous forme d’un tableau
Excel et serviront pour l’analyse informatique.
b) Traitement du signal et normalisation de l’intensité des spots
Comme l’illustre la
Figure 3, l’intensité du signal obtenu pour chaque spot est
variable. L’intensité du signal dépend de plusieurs paramètres dus à la
manipulation. Pour permettre de comparer deux expériences, il est impératif de
normaliser les données. En effet, l’intensité d’un spot peut varier d’une
expérience à l’autre en fonction de l’efficacité du marquage de l'ADN
génomique, du temps d’exposition…
La normalisation
s’effectue sur la moyenne des valeurs des taches pour lesquels il y a un signal
d’hybridation. En effet, le nombre de taches ne donnant pas de signal est
variable d’une souche à l’autre, et une normalisation seulement basée sur la
moyenne de l’ensemble des points entraînerait une erreur significative.
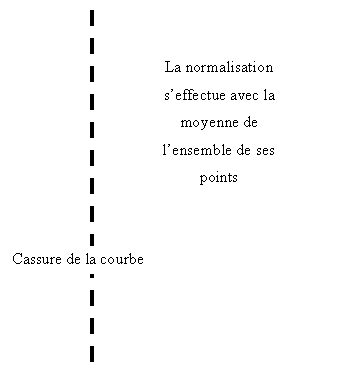
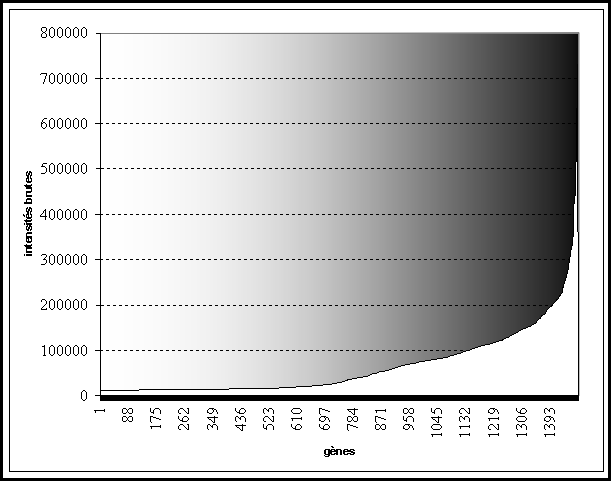
Figure 6 : Intensité brute classées par ordre croissant resultant
de l'hybridation d'une souche de L. monocytogenes provenant de Helsinki
A partir des
résultats de quantification avec Array-vision®, une courbe correspondant à la
valeur brute quantitative de chaque spot est tracée en fonction de cette
valeur. Tous les gènes situés avant la cassure de la courbe ont une valeur non
significative car ils sont situés dans le bruit de fond. La moyenne de
l’intensité des spots est calculée à partir de ce point. Les valeurs sont
finalement normalisées en divisant la valeur quantifiée par cette moyenne (Voir
Figure 6).
c) Membrane virtuelle de références et détermination de valeur
« ratio »
L’intensité
d’hybridation donne une valeur relative et pour définir si un gène est présent
ou absent, il est nécessaire de diviser ces valeurs normalisées par une valeur
de référence normalisée obtenue avec un ADN génomique qui contient ce gène. On
obtient alors les valeurs ratio. L’intensité normalisée de référence pour un
gène est celle obtenue par hybridation avec les ADNg des souches matrices de la
puce utilisés pour faire les PCR.
Pour déterminer ces
valeurs de référence, les hybridations avec les ADNg de ces souches matrices
ont été réalisées en triplicata. Une moyenne des trois valeurs normalisées est
effectuée et une membrane virtuelle de référence est réalisée en combinant les
valeurs d’hybridations des souches matrices.
d) Notion de seuil et d’ambiguïté
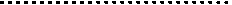

Zone d’ambiguïté : le seuil de coupure est définit
à 30%
|
|





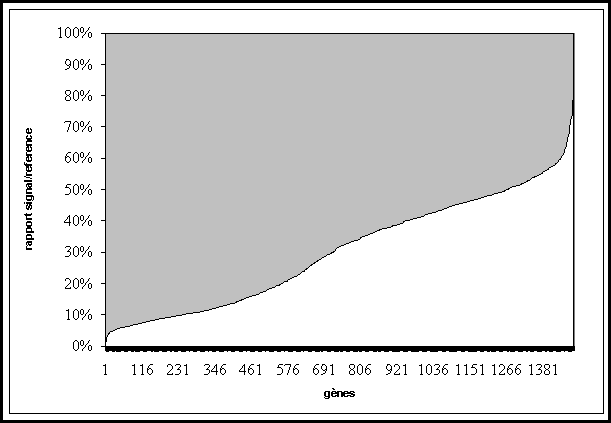
Figure 7 : Rapports classés par ordre croissant
des intensités normalisées entre le signal et la référence en fonction des
gènes (valeurs ratio), ADNg d’une souche L. monocytogenes en provenance de
Helsinki hybridées sur la puce de 2eme génération
Le point critique
dans l'analyse est de prédire à partir des valeurs ratio si un gène est présent
ou absent. Expérimentalement un seuil de coupure est défini par rapport aux
valeurs « ratio » qui permet de minimiser le nombre de faux positif
sur la base de l'hybridation avec les génomes de séquence connue. Les valeurs
« ratio » seront comparées à ce seuil de coupure et permettront de
prédire l’absence ou la présence du gène au sein de l’ADNg. (Voir Figure 7)
Les signaux obtenus
ne sont pas tous tranchés. Comme le montre la Figure
7, les valeurs des intensités de certains signaux se
situent dans une zone d’ambiguïté où il est difficile de binariser le signal.
D'une expérience à l'autre ce signal sera au dessus ou au dessous du seuil.
Les valeurs des
ratio d’une hybridation de l’ADNg de EGDe ont été classé par ordre croissant et
séparé selon si le gène est effectivement présent ou absents dans le génome. Il
peut aussi exister des faux« négatif » c'est-à-dire que ces signaux auraient
du être positif.
A l'aide des
macro-arrays, on peut caractériser les génomes des souches par la présence ou
non des gènes. L’hybridation de l’ADNg de plusieurs centaines de souches de
listéria d’origines diverses (environnementales, cliniques ou portage) va
permettre de constituer une base de données très importante mais difficilement
interprétable. C’est pour cela que plusieurs algorithmes ont été appliqués pour
être intégré à travers différents logiciels. Ces logiciels, comme J-Express,
vont permettre de regrouper hiérarchiquement (classification hiérarchique) les
gènes recherchés et les souches entre elles. Plusieurs études comparatives
entre les souches sont rendues possibles grâce au regroupement hiérarchisé
comme, par exemple, l’étude des souches d'origine clinique comparées aux
souches environnementales.[9]
a) Traitement des données par J-Express
Après avoir défini
le seuil de coupure permettant d’interpréter le signal pour une présence ou une
absence de gènes, une matrice binaire, regroupant les gènes en fonction des
souches, est créée. Cette matrice est incorporée dans le logiciel J-Express qui
permet de réaliser le regroupement hiérarchisé par UPGMA (Unweighted Pair Group
Method with Arithmetic mean).
Figure 8 : Interface du
logiciel J-Express
J-Express réalise
un double regroupement hiérarchique où à la fois les gènes et les souches sont comparés.
Par l’intermédiaire d’un module graphique, J-Express construit un arbre où les
souches en abscisse d’une part et les gènes en ordonnées d’autre part sont
clustérisées regroupés. La présence de gène dans la souche est symbolisée par
un pixel rouge. Une difficulté liée à la classification hiérarchique et l'utilisation
des scores binarisés. Cette classification est donc très sensible aux scores
des valeurs proches du seuil de binarisation.
b) « X strains » : une solution alternative pour regrouper
les souches
L’analyse d’une
membrane de typage implique une étape de décision pendant laquelle
l’utilisateur doit décider si le gène, en fonction du signal et d’un seuil, est
présent ou absent. Cette décision va influencer par la suite le regroupement
hiérarchisé des souches étudiées entre elles.
« X-strains »
est un progiciel développé par Christophe Rusniok de l'Unité GMP en complément
de la méthode de classification hiérarchique dans le but de retirer cette étape
de « binarisation » lors du traitement du signal des puces de typage.
Il permet de former des groupe de souches tout en connaissant les distances
(nombre de signaux comparés pour un gène significativement diffèrent) entre les
souches. La méthode d’analyse est basée sur la comparaison souche par souche
(une souche contre toutes les autres souches). Les souches qui sont identiques
par cette méthode vont être regroupées. Cette nouvelle approche devrait
permettre de définir des groupes des souches qui se ressemblent et de
définir les souches qui sont proches ou identiques.[24]
Malgré l’intérêt
des puces d’hybridations ADN/ ADN sur les autres techniques de typage par l’apport
de connaissances génomiques, l'expérience acquise sur les puces de 1ère
et 2ème génération a mis en évidence différentes difficultés (Tableau 2). Cette technique est limitée par le bruit de fond
biologique et technique ainsi que par le pouvoir discriminatoire et la
représentativité des gènes sélectionnés pour construire la puce.
Tableau 2 : Liste des difficultés rencontrées
|
|
Difficultés rencontrées
|
Solutions envisagées
|
|
1
|
Certaines souches hybrident trop faiblement pour donner un
signal interprétable malgré plusieurs tentatives.
|
Choix de la méthode d’extraction d’ADN donnant les meilleurs
marquages.
|
|
2
|
Après normalisation, 17% en moyenne des signaux ont des
valeurs ratio (comprise entre 0,2 et 0,4) proche du seuil de coupure de 0,3
et donc difficile à interpréter.
|
Améliorer le rapport signal/ bruit en choisissant les
meilleurs sondes.
|
|
3
|
Après l’analyse par regroupement hiérarchique, le typage des
souches peut être effectué. Cependant l’analyse de 739 gènes à travers le
logiciel J-express montre que 27% des gènes apporte peu de poids en étant
soit présents ou absents dans plus de 95% des souches.
|
Analyser la valeur de chaque gène dans l’information qu’il
apporte réellement dans la discrimination des souches entre elles.
|
|
4
|
La puce à ADN se base essentiellement sur le génome de L. monocytogenes EGDe de sérovars
1/2a. Or, celle-ci clustérise par cette technique avec les L. monocytogenes de sérovars
1/2c. Par conséquent, cette souche n’est pas forcément représentative de la
diversité des souches 1/2a
|
Intégrer à la nouvelle puce des gènes provenant d’une L. monocytogenes de sérovars 1/2a
aspécifiques de EGDe.
|
|
5
|
Bien que les phages (non présent sur la dernière puce mise au
point) n’apporte que peu d’information sur les Listeria du point de vue
fonctionnelle, ils permettraient tout de même d’apporter un poids
supplémentaires dans l’interprétation de la variabilité du génome de
Listeria.
|
Réintégrer les gènes de bactériophages qui ont pu être écarté
entre la puce 1 et la puce 2.
|
|
6
|
Certains gènes sont redondants, c’est-à-dire qu’ils
appartiennent à un groupe de gènes qui donnent le même signal.
|
Choisir au sein de ces clusters les sondes qui donnent le
meilleur signal.
|
a)
Bruit de fond
La principale limite de cette technique d’hybridation
moléculaire réside dans l’interprétation du signal attestant la présence ou
l’absence de la séquence nucléique recherchée. Le bruit de fond, d’origine
technique et biologique, est une des causes principales de cette limite.
En effet, le bruit
de fond résultant en partie au moins d’hybridations croisées (causés par la
présence de gènes paralogues) peut fausser l’interprétation du signal et
générer de faux positifs dans le cas d’hybridations croisées et de faux négatif
dans le cas d’un bruit de fond trop élevé. Les hybridations croisées sont
variables entre les souches et les gènes. Le bruit de fond est aussi généré par
le matériel utilisé lors de la manipulation. : C’est le bruit de
technique. Les membranes de nylon et le scanner Typhoon® sont responsables de
ce bruit de fond technique. Dans le cas de la méthode X-strains, l'importance
des comparaisons de souches proches permet de réduire l'influence du bruit de
fond biologique.
b) Pouvoir discriminatoire d’un gène déposé
Malgré le choix des
gènes « déposés » spécifiques de quatre génomes de
listéria, certains gènes ayant une distribution homogène, c’est-à-dire présents
ou absents dans toutes les souches, apportent peu de poids quant à la
différentiation des souches par classification hiérarchique.
Il semble important
de noter que la mise en évidence de gènes de phage peut permettre de mieux
comprendre la relation entre deux souches similaires qui sont reconnues comme
différentes par une analyse en champs pulsé. Enfin, Il serait nécessaire de
diversifier la puce en complétant les sondes définies à partir du génome de la
souche EGDe par des gènes spécifiques de la souche L. monocytogenes
de sérovars 1/2a séquencée par l’institut de génomique TIGR[22].
Le but de la mise au point d’une puce de 3eme génération est
de proposer un outil de typage plus efficace en mettant à profit l’expérience
sur les deux autres puces avec moins de contraintes en terme d’analyse (réduction
du nombre de taches à analyser) avec des signaux plus tranchés sans ambiguïté
tout en gardant la représentativité des gènes à travers les diffèrent génomes
de listéria séquencés. Il s’agira en particulier de résoudre les difficultés
énoncées dans le Tableau
2.
Cette mise au point a été effectuée par plusieurs
étapes :
Ø
Une synthèse des résultats des
puces de première et deuxième génération.
Ø
Une sélection des gènes présentant
un potentiel discriminant important et sans ambiguïté dans le but de réduire le
bruit de fond biologique.
Ø
Une étude sur influence des
différentes étapes des développements et d’utilisation des puces à ADN
(produits PCR, extraction ADN, température d’hybridation, membrane de nylon,
concentration de l’ADN génomique matrice pour l’amplification des sondes) dans
le but de réduire le bruit de fond technique
À partir des données obtenues par les puces de 1ere et 2eme
génération et des souches matrices, une sélection de gènes est effectuée pour
la création de cette nouvelle puce dite de « troisième génération »
(puce 3G). Les critères de sélection sont définis de manière à écarter les
gènes qui présentent une hybridation croisée ou apportant peu d’information.
Les fragments spécifiques des gènes sélectionnés seront alors amplifié par PCR.
Les amplicons seront vérifiés par électrophorèse sur gel et par séquençage
avant d’être déposés sur la membrane.
Ensuite, le protocole d’analyse de la puce seront décrit
pour le logiciel Array-vision® de tel manière à utiliser la nouvelle puce en
routine. Les souches matrices ainsi que les souches références seront hybridées
sur la nouvelle puce pour construire la première base de données.
Les souches matrices (Voir Tableau
3) sont utilisées pour amplifier par PCR l’ADN des
gènes «à déposer », et pour les hybridations des puces permettant de réaliser
la membrane de référence virtuelle.
Tableau 3 : Souches matrices utilisées pour le
développement de la nouvelle puce
|
Souche matrice
|
sérovars
|
indicatif gènes
correspondant
|
|
L. monocytogenes
EGDe
|
1/2a
|
Lmo
|
|
L. monocytogenes (CLIP80459)
|
4b
|
Lm4b
|
|
L. monocytogenes (CLIP90602)
|
1/2b
|
Lm12b
|
|
L. innocua (CLIP11262)
|
6a
|
Lin
|
|
L. monocytogenes TIGR F6854
|
1/2a
|
Tg12a
|
|
L. monocytogenes TIGR F2365
|
4b
|
Lm4b-T
|
*CLIP : Collection Listéria Institut Pasteur
Pour construire la base de l’arbre de regroupement
hiérarchique, nous avons choisi d’hybrider les souches de références de
différents sérovars de toute les espèce du genre Listeria. Ces souches
sont la référence pour leur espèce et leur sérovar. Pour tester la performance
et le pouvoir discriminatif de la nouvelle puce, d’autres souches seront
hybridées sur la nouvelle puce. 10 souches du CDC responsables d’épidémie aux
Etats- Unis[10], 4 souches isolées en Finlande responsable d’épidémie,
ainsi que 3 souches isolées en France dont 2 proviennent des industries
alimentaires (Voir Tableau
4).
Tableau 4 : Descriptions des souches à hybrider
sur le la puce 3G
|
Souches de références
|
|
Autres souches hybridée
sur la puce 3G
|
|
Espèce
|
sérovars
|
CLIP
|
|
dénomination
|
sérovars
|
information°
|
|
L. monocytogenes
|
1/2a
|
74902
|
|
CDC de 1 à 10
|
4b
|
Souches de L. monocytogenes du control
desease center of Atlanta
|
|
1/2b
|
74903
|
|
3H
|
1/2c
|
Souches L. monocytogenes provenant de
l’université de Helsinki
|
|
1/2c
|
74904
|
|
5H
|
1/2c
|
|
3a
|
74905
|
|
9H
|
1/2a
|
|
3b
|
74906
|
|
12H
|
1/2b
|
|
3c
|
74907
|
|
CLIP933666
|
1/2a
|
L. monocytogenes
Portage femme enceinte 1991
|
|
4a
|
74908
|
|
CLIP93149
|
12a
|
L. monocytogenes
Filière Lait Epoisse 2001
|
|
4b
|
74910
|
|
CLIP93655
|
1/2a
|
L. monocytogenes Entremont environementale
|
|
4c
|
74911
|
|
|
|
|
|
4d
|
74912
|
|
|
|
|
|
7
|
74917
|
|
|
|
|
|
3c
|
85412
|
|
|
|
|
|
L. innocua
|
6a
|
74915
|
|
|
|
|
|
6b
|
74916
|
|
|
|
|
|
4ab
|
74909
|
|
|
|
|
|
L. ivanovii
|
5
|
74914
|
|
|
|
|
|
L. welshimeri
|
6b
|
74020
|
|
|
|
|
|
L. seeligeri
|
1/2bnc
|
86579
|
|
|
|
|
Les données utilisées pour la sélection des gènes à déposer
sur la puce 3G sont d’une part les tableaux regroupant les valeurs ratio de la
puce 1 et de la puce 2 et d’autre part l’accès aux génomes des souches matrices
sur le site de « génolist »
de l’Institut Pasteur sous la forme de fichier IPF et
sur le site de TIGR.
Un tableau de comparaison des quatre génomes des souches
EGDe, L. innocua, L. monocytogenes 4b et 4b TIGR a été
préalablement effectué. Le génome de L. monocytogenes 1/2a TIGR a
été comparé aux quatre génomes précèdent par l’intermédiaire d’un programme
développé par Christophe Rusniok basé sur l’algorithme de Blast.
La réaction de polymérase en chaîne (PCR) pour obtenir des
amplicons de gènes est une étape cruciale dans la réalisation des membranes
d’hybridation ADN/ADN. C’est pour cela qu’il est important d’en étudier son
influence pour en contrôler ses conséquences sur le signal de la puce. Cette
étude se portera sur la concentration d’ADN génomique à amplifier et sur
l’influence de la qualité de la PCR sur le bruit de fond.
Les oligonucléotides correspondant aux souches des génomes
séquencés par l’unité GMP ont été préalablement définis et sont accessibles
directement à partir de la base de données du laboratoire. En ce qui concerne,
les génomes des souches de TIGR, les oligonucléotides ont été dessinés avec le
logiciel Primer 3.
Les oligonucléotides sont dessinés de manière à ce que les amplicons des gènes
ont une taille comprise entre 400 et 600pb.
Les réactions d’amplification par PCR « en masse »
pour la préparation de la macro-array sont réalisées en plaque 96 puits. Pour
chaque réaction de PCR, un mélange réactionnel est réalisé (Voir Tableau 5)
Tableau 5 : Mélange
réactionel pour la PCR et programme utilisé pour le thermocycleur (GeneAmp)
|
Réactif (Applied)
|
Volume
|
Concentration
|
|
temps
|
températures
|
Nbre cycles
|
|
ADN génomique dilué
|
1 ml
|
A déterminer
|
|
5’
|
94°C
|
1 cycle
|
|
dNTP (10 mM)
|
2 ml
|
10 mM
|
|
30’’
|
94°C
|
30 cycles
|
|
Mgcl2 (25 mM)
|
8 ml
|
25 mM
|
à
|
1’
|
50°C
|
|
tampon 10X
|
10 ml
|
10X
|
|
1’
|
72°C
|
|
Taq polymérase
|
0,5 ml
|
5UI/ml
|
|
7’
|
72°C
|
1 cycle
|
|
eau distillée
|
qsp 90 ml
|
|
∞
|
4°C
|
-
|
Dans chaque puits de la plaque PCR, il faut mettre 10 ml d’amorce
(10 mM)
spécifiques de la sonde à amplifier et 90 ml de mélange réactionnel sont ajoutés respectivement. Le
volume de la réaction est donc de 100 ml. Le programme de la réaction d’amplification est aussi
décrit dans le Tableau 5.
Pour vérifier l’amplification,
la concentration et la spécificité des produits de PCR attendu, ils sont
déposés sur gel 1% d’agarose en tampon TBE 1X (Tris Borate EDTA). La révélation
s’effectue avec du BET.
Pour chaque gène, 10 ml de produit de PCR et 4 ml de tampon de charge sont déposés. En parallèle des
produits de PCR, 3 ml de
marqueur de poids moléculaire (Smart ladder) sont déposés afin de pouvoir
déterminer la taille de chaque amplification. Après la migration, le gel est
exposé par exposition aux rayons UV pour révéler l’ADN marqué au BET.
Après l’amplification et la
vérification, les amplicons de gènes sont déposés sur une membrane en nylon
pour constituer une macro-array.
a)
Principe de la fixation de
fragment d’ADN sur membrane
La fixation de l’ADN sur un support se fait soit directement
par interaction entre les groupes phosphates de l’ADN et les cations de la
membrane (membranes chargées), soit par irradiation aux UV ou cuisson sous
vide, ce qui fixe de manière covalente l’ADN à la membrane (membrane non
chargée). La dénaturation de l’ADN pour le rendre simple brin se fait par
traitement alcalin. Dans le cas présent, des membranes chargées positivement
seront utilisées. Les produits de PCR doubles brins qui sont déposés se fixent
donc par covalence et sont dénaturés par une solution de NaOH/NaCl et par un
traitement aux UV.
b) Caractéristiques des membranes et pilotage du robot Qpix
Les membranes utilisées ont une
dimension de 22,2 x 22,2 cm. Avec une membrane de cette taille, on obtient 6
« petites » membranes, chacune contenant l’ensemble des amplicons de
gènes. Les amplicons de gènes sont déposés en duplicata sur la membrane à
l’aide du robot Qpix (Genetix).
Figure 9 : Interface du logiciel de pilotage « Gridding »
du Qpix de Genetix
Le logiciel « Gridding » (voir Figure 9) permet de définir les paramètres indiqués dans le Tableau 6 avec lesquels la membrane à haute densité sera
réalisée. Dans ce logiciel, tout d’abord le nom de la feuille de route est
choisit, le type de plaques dans lesquelles se trouvent les gènes à déposer
(plaque 96 ou 384 puits), le support de la membrane (bloc ou boîte), le nombre
de champs à réaliser sur la membrane, le praton (nombre de fois et positions où
sera déposée une même sonde), les différents temps et nombre de fois ou les
aiguilles seront lavées dans l’éthanol et dans l’eau, et la tête du robot à
utiliser (96 ou 384 aiguilles).
Tableau 6 : Paramètres de pilotage du Qpix pour le
dépôt des sondes sur la macro-array
|
Paramètres
|
|
|
Tête
|
384 PIN Gravity gridding
|
|
Plaque source
|
Abgene plaque 384 well
|
|
Spot par gène
|
2 réplicats
|
|
Lavage : éthanol/eau
|
4/4
|
|
Séchage
|
5 secs
|
|
Distance entre deux patterns
|
2500 mm
|
|
Nombres de dépôts par spot
|
5
|
|
Temps sur membrane (aiguilles)
|
2000 ms
|
|
Temps dans un puit (aiguilles)
|
1000 ms
|
L’ADNg est dénaturé par la
chaleur (99°C) pour obtenir de l’ADN simple brin. Le marquage est réalisé par
la méthode de l’initiation aléatoire. Cette méthode est basée sur un mélange de
tous les hexanucléotides possibles pouvant s’hybrider sur n’importe quel partie
de l’ADN. Le brin complémentaire est synthétisé à partir de ce mélange par
l’intermédiaire de l’enzyme Klenow. Les quatre désoxynucléotides dont un seul
est marqué radioactivement par le 33P (il s’agit du dCTP) sont
incorporé dans la synthèse du nouveau brin d’ADN.
a)
Marquage de l’ADNg
10 ml
d’ADN (5 ng/ml)
sont dénaturés en thermocycleur pendant 10 minutes à 99°C. Les tubes sont
immédiatement mis dans la glace pour éviter la réassociation des deux brins. Pour
effectuer le marquage, on utilise le kit « Random Priming DNA
Labeling » de chez Roche. Aux 10 ml d’ADN dénaturé, les réactifs suivants sont
ajoutés :
Ø
1 ml dGTP (0,5 mM)
Ø
1 ml dTTP (0,5 mM)
Ø
1 ml dATP (0,5 mM)
Ø
2 ml mélange réaction (tampon +
hexamères)
Ø
5 ml 33P dCTP
Ø
1 ml enzyme de Klenow
Le volume final de la réaction de marquage est de 21 ml. La
réaction de marquage se fait durant 45 minutes à 37°C en thermocycleur. La
réaction est arrêtée en ajoutant 2 ml
d’EDTA 0,2M pH=8
Remarque : l’ajout d’EDTA 0,2M pH=8 n’est pas
nécessaire si l’ADN est purifié dès la fin du marquage.
b) Purification de l’échantillon
Les ADN néosynthétisés à
partir de l’ADNg sont purifiés et séparés des nucléotides, marqués ou non, non
incorporés par le kit Qiaquick Nucleotide Removal. Les nucléotides non
incorporés, sont éliminés pour éviter un signal aspécifique.
L’ADN est purifié sur colonne comprenant une membrane qui
retient des fragments d’ADN ayant une taille supérieure à 17 bases. La liaison
de l’échantillon d’ADNg à la membrane est facilitée à forte concentration en
sel. Après deux lavages successifs pour éliminer tous les contaminants, l’échantillon
est éluée en diminuant la concentration en sel et en augmentant le pH.
10 volumes de tampon PN sont ajoutés à un volume d’ADN
marquée. La réaction de marquage se faisant dans un volume de 21 ml,
210 ml de
tampon PN sont ainsi ajoutés. Les 231 ml de solution sont déposés sur la colonne puis centrifugée
une minute à 6000 rpm. L’ADN est retenue sur la colonne. La colonne est placée
sur un nouveau tube Eppendorf et lavée avec 500 ml de tampon PE. Après une minute de centrifugation à
6000 rpm et après avoir éliminé le filtrat, un deuxième lavage est effectué
avec à nouveau 500 ml de
tampon PE. Après retrait du filtrat, la colonne est centrifugée 1 minutes à
13000 rpm avant d’être placée sur un tube Eppendorf afin d’éluer l’échantillon
avec 200 ml de
tampon d’élution EB. L’élution s’effectue par une centrifugation d’une minute à
13000 rpm.
c)
Dénaturation de l’ADNg marquée et purifiée
Après avoir purifié l’échantillon, on le dénature par passage
à 99°C afin d’avoir de l’ADN simple brin pouvant s’hybrider avec les ADN déposés
sur la membrane en cas de complémentarité. En éluant par 200 ml de
tampon EB, l’échantillon est dénaturé pendant 10 minutes à 99 C puis
laissé à 4°C avant d’être ajouté à la solution d’hybridation.
Avant d’hybrider l’échantillon
d’ADNg sur la membrane, cette dernière est saturée avec 10 mL une solution
d’hybridation (Voir Tableau
7) contenant du Denhardt’s (Sigma) et de l’ADN qui
provient de sperme de saumon qui se fixe sur la membrane « vierge »
d’ADN. Cette étape permet d’éviter la fixation non spécifique de l’ADN sur la
membrane. Après avoir préchauffé la solution d’hybridation à 60°C, la membrane
est préhybridée une heure à 60°C dans 10 ml de cette solution sous rotation
dans un four à hybridation.
Tableau 7 : Solution d’hybridation pour une membrane
|
SSPE 10X
|
7,5 ml
|
|
Composition SSPE 10X
|
|
SDS 10%
|
3,5 mL
|
|
NaCl 5M
|
360 mL
|
|
Denhardt’s 50X
|
0,3 mL
|
|
EDTA 0,5M pH=8
|
20 mL
|
|
ADN saumon 10µg/µL
|
3µL
|
|
NaH2PO4 1M
|
86,7 mL
|
|
H2O
|
4,5mL
|
|
Qsp 1L d’eau distillée
|
a)
Hybridation de l’ADNg marqué aux
cibles d’ADN immobilisées (sondes) sur membrane
La réaction d’hybridation met en jeu deux chaînes d’ADN
simple brin (la cible et l’échantillon). Pour choisir la température
d’hybridation, il faut tenir compte de la température de fusion qui est la plus
importante, de la taille de la cible, sa teneur en GC%, la succession de 3A,
3T, 3C et 3G, la concentration en ions monovalents et en agents déstabilisant
(formamide ou urée).
L’ADNg marquée et dénaturée est ajoutée à 5 ml de solution
d’hybridation. Cette solution est incubée sur la membrane après avoir éliminé
la solution de préhybridation. L’hybridation se fait sur la nuit à 60°C sous
rotation.
b)
Lavages de la membrane
Une fois l’hybridation terminée, la solution d’hybridation
contenant l’excès d’échantillon est
éliminée et la membrane est ensuite lavée par plusieurs lavages successifs qui
permettent d’éliminer l’excès d’échantillon ou d’ADN qui se sont hybridées de
manière aspécifique. Ces lavages sont effectués à la même température que
l’hybridation. Ainsi, on est à forte astringence ce qui permet de conserver les
hybridations spécifiques et de déstabiliser les liaisons faibles entre deux
séquences d’ADN non parfaitement complémentaire.
La membrane est lavée deux fois trois minutes à température
ambiante sous agitation dans une solution de lavage (50 ml SSPE 10X, 20ml SSC
2X qsp 1L d’eau distillée). La solution est ensuite incubée à 60°C afin de
réaliser deux lavages de 20 minutes sous agitation à 60°C. Après les différents
lavages, la membrane est mise entre deux feuilles plastiques qui sont ensuite
soudées tout en vérifiant bien qu’il n’y ait ni bulles ni plis. La membrane
ainsi « plastifiée » est de nouveau soudée dans un second plastique
pour éviter de contaminer l’écran qui sera déposé sur la membrane dans la
cassette. L’écran est exposé à la lumière pour être totalement désactivé avant
d’être exposé sur la membrane. Ce dernier capte l’énergie de la radioactivité
provenant de la membrane. L’écran est exposé deux jours sur la membrane. A la
fin de la manipulation, par exposition à la lumière, l’écran est effacé pour
être réutilisé avec une nouvelle membrane.
c) Révélation de l’hybridation
L’écran est scanné grâce à un
PhosphorImager (Typhoon, Amersham) qui détecte l’intensité de la radioactivité
des différents spots mais aussi de la membrane émise par l’écran. Le balayage est
réalisé avec une taille de pixel de 100 microns. Plus la taille de pixel est
faible plus la résolution est haute.
d)
Déshybridation de la membrane
Une fois que la membrane est scannée, elle peut être
déshybridée et réutilisée pour une nouvelle hybridation. Chaque membrane peut
être utilisée 4 à 5 fois. La technique de déshybridation à la soude est
utilisée : la membrane est placée dans une solution de NaOH 0,5M
durant 30 minutes sous agitation. Elle est ensuite rincée à l’eau distillée,
trempée rapidement dans une solution de SSC2X et conservée à -20°C.
|
Remarque : gestion
des déchets radioactifs et surveillance des contaminations radioactives
|
|
Les manipulations contenant
une étape de manipulation d’isotope radioactif sont soumis à des contraintes
de sécurité. Un poste dans le laboratoire doit uniquement servir à cet effet.
Ce poste est décontaminé après chaque manipulation. Chaque manipulateur
reçoit une formation de sensibilisation à la radioactivité. Tous les déchets
radioactifs (solide et liquide) sont triés pendant la manipulation.
L’Institut Pasteur prend en charge la gestion et la décroissance des déchets
radioactifs. Ainsi peu de laboratoires ont accès aux manipulations contenant
de la radioactivité.
|
Une fois que les membranes ont été scannées, elles sont
analysées grâce au logiciel Array-vision®. Plusieurs étapes sont nécessaires
avant de pouvoir faire l’analyse des nouvelles puces :
Ø
Construire la nouvelle membrane de
référence
Ø
Définir le nouveau protocole
d’analyse dans Array-vision
Ø
Traiter le signal (comme indiqué
en I.D.1) pour construire une nouvelle base de données
Plusieurs simulations « in silico » ont été
nécessaires avant d’adopter une stratégie pour réaliser une sélection des gènes
à déposer sur la membrane. Au cours de cette sélection, il faut aussi veiller à
la représentativité des gènes dans le but de garder le pouvoir discriminatif de
la puce « biodiversité ».
Pour réaliser efficacement une sélection des gènes provenant
des puces de 1ere et de 2eme génération, une étape in silico a été
nécessaire par l’intermédiaire de simulation sur le logiciel J-express après
analyse de l’information sur le tableur Excel afin de vérifier à la fois le
potentiel discriminatoire mais également la représentativité des gènes choisis
pour la puce 3 G.
La sélection des gènes a été basée sur plusieurs critères
d’analyse de données à la fois biologiques et qualitatifs :
Ø
Les sondes sélectionnées doivent
donner un signal non ambigu, car les sondes générant des hybridations croisées
sont responsables des signaux ambigus et conduisent à un biais dans l’analyse
des résultats de typage
Ø
L’ensemble de sonde sélectionnée
doit avoir le même pouvoir discriminatoire que celui des puces construites
auparavant.
Ø
Les sondes sélectionnées sont
aussi celles qui sont importantes/impliquées dans l’interaction de L .monocytogenes
et l’environnement et les facteurs de virulence connues (motif LPXTG, famille
des internalines, …).
Ø
Nous effectuons aussi le choix de ne
prendre qu’une sonde pour des gènes organisés en opéron qui donne une fréquence
de signaux identiques
Ø
Les gènes présents ou absents dans
toutes les souches ne sont pas discriminants pour permettre de mettre en
évidence des sous-groupes au sein des sérotype de L. monocytogenes.
L’utilisation de J-express
passe tout d’abord par une étape sur le tableur Excel de Microsoft. La
comparaison de l’intensité du signal d’un gène en fonction des différents
critères est effectuée par l’intermédiaire d’un test logique de condition
« if then else » (voir Figure
10).
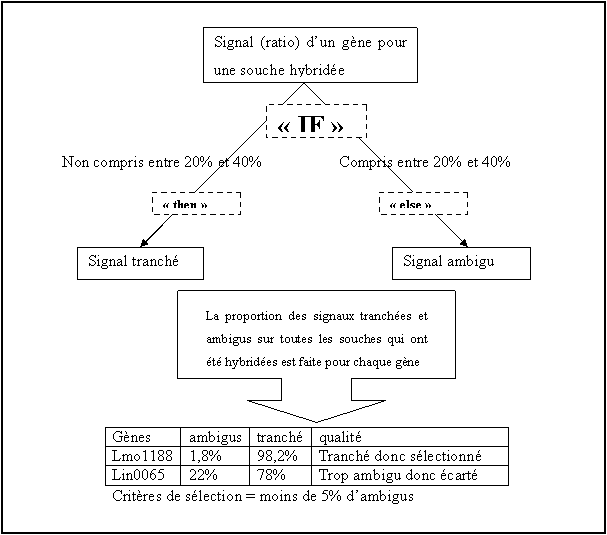
Figure 10 : Organigramme décisionnel "if then
else"
Les critères de sélection sont définit en fonction de la
valeur du ratio et de la répartition du signal dans toutes les souches testées.
Un signal est dit trop ambiguë lorsque la valeur de son ratio se situe entre
0,2 et 0,4 dans plus de 5 % des souches testées. Les signaux présentant des
caractéristiques de manière générale trop ambiguë sont écartés de cette
manière. Cette technique sera appliquée sur toutes les souches mais également à
l’intérieur des souches du même sérovars. Dans les deux cas, 200 gènes seront
sélectionnés.
Remarque : L’hybridation de EGDe avec la puce de 2eme
génération produit 12,7% de signaux ambigus.
b) Ecarter les gènes qui apporte peu d’information pour discriminer les
souches
De la même manière que
précédemment à l’aide d’un test logique sur le tableur Excel, les gènes donnant
des signaux « identiques » pour toutes les souches seront écartés. Un
signal est dit identique dans toutes les souches si l’hybridation donne la même
réponse dans plus de 95% des souches.
c)
Écarter les gènes ortho logues
Par l’intermédiaire du développement d’une macro (Tableau 8) un test de comparaison de génome est effectué pour
retirer les sondes en « doublon » provenant de deux génomes.
Tableau 8 : Macro sub test() applicable sur les
logiciels Office Mac OS
|
Sub test()
|
Definition de la macro
|
|
For Each o In Range("B1:B" &
Range("B65000").End(xlUp).Row)
|
Pour tout les élément de
la colonne B on définit une valeur « o »,
|
|
Set FD = Range("D1:D" &
Range("D65000").End(xlUp).Row).Find(o.Value)
|
Pour tout les élément on
définit un variable FD définit par la recherche des valeurs « o »
|
|
If Not FD Is Nothing Then
|
Si La variable FD existe
réellement
|
|
z = FD.Address
|
On définit une valeur “z”
|
|
o.Offset(0, -1).Value = Range(z).Offset(0,
-1).Value
|
« z » est alors
égale à la valeur de la colonne de gauche de D et sera inscrit dans la
colonne à gauche de B
|
|
End If
|
Fin de la condition
|
|
Next
|
Cette opération est
réalisée pour toutes les cellules
|
|
End Sub
|
Fin de la macro
|
Cette macro va permettre de comparer deux colonnes dans un
tableau Excel et lui attribué une valeur. Ces deux colonnes peuvent être par
exemple les gènes des génomes de EGDe et de L. innocua. Cependant
il faut utiliser un feuillet où la comparaison des génomes avait déjà été
effectuée par Blast pour pouvoir adapter cette information a la liste gène de
la puce dans une autre colonne.
La Figure
11 montre en colonne C et D les génomes de L. monocytogenes
EGDe et L. innocua. Ces génomes ont été préalablement comparés par
Blast c'est-à-dire que les gènes se trouvant sur la même ligne sont orthologues.
On souhaite rechercher dans les gènes amplifiés à partir de L. innocua
présent sur la puce (colonne B) les gènes lmo en colonne A (L. monocytogenes
EGDe) orthologues. Le gène lin0059 va être recherché dans la colonne D (voir
chemin 1 Figure 11). La macro trouve la correspondance dans la colonne C
avec le gène lmo0066 et va le reporter en colonne A en face du gène voulu (voir
chemin 2 Figure 11). En revanche, lorsque que la macro ne trouve pas de
correspondance, elle n’inscrit rien en face des gènes recherchés et passe à la
ligne suivante (voir chemin 3 Figure
11).

Figure 11 :
Illustration de l'utilisation de la macro « subtest » dans la cas
d'une recherche rapide de gènes lin dans le génome
de EGDe
Cette macro va permettre de comparer rapidement en un seul
clic plusieurs listes de gènes. Elle permettra d’écarter par exemple les gènes
en doublon sur la puce ou bien de manière plus pratique de faire les
correspondances entre le système d’annotation progressif du génome (fichier
IPF) et les annotations finales. Cette macro permet également de mettre en
relation plus facilement les données obtenues avec les anciennes puces et la
puce actuelle pour des simulation de groupage de souches in silico sur
J-Express.
Trois grandes étapes de sélection des gènes ont été
effectuées dans le but de réduire le nombre de gènes présents sur la puce tout
en préservant l’information sur la virulence et enfin diversifier la puce par
l’ajout de nouveau gènes.
La Figure
12 illustre la procédure de sélection des gènes à partir
des données des précédentes puces et des données des génomes de TIGR. Pour
toutes ces étapes de sélection in silico, la macro « subtest »
a été utilisé en complément des fonctions de base de Excel.
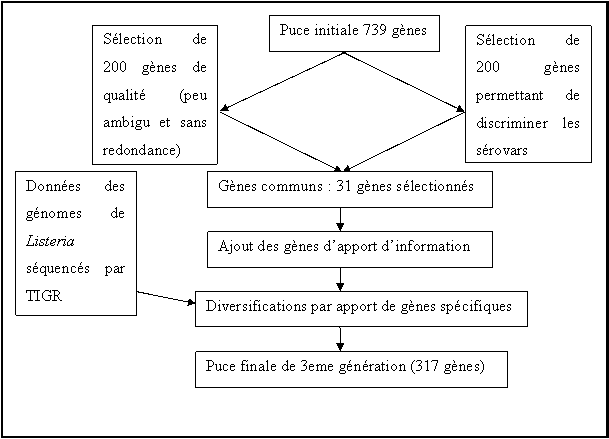
Figure 12 :
Présentation des étapes de selection des gènes
Dans un premier temps, à l’aide de critères sélectifs, une
puce virtuelle a été construite sur la base d’une trentaine de gènes seulement.
Cette puce virtuelle d’une trentaine de gènes est un squelette de la future
puce. Néanmoins les gènes seront sélectionnés de manière à ce que cette puce
soit assez discriminante pour différencier les sérovars. Pour cela 200 gènes
seront sectionnés par des critères de qualité (absence d’ambiguïté et une
fréquence de signal différentes en fonction des sérovars) et 200 gènes seront
sélectionnés dans leur pouvoir à discriminer les sérovars entre eux (Voir Figure 10 et cf. III.A.1.a)). Les gènes communs à ces deux ensembles de gènes
constituent le squelette de la future puce.

Figure 13 : Analyse en Composantes Principales d’une
simulation « in silico », d’hybridation de 10 souches de référence
avec les 31 sondes choisis dans la Figure 12
La Figure
13 illustre l’Analyse en Composante Principale d’une
simulation avec les 31 sondes choisis comme « individus » et 10
souches de référence comme « variable ». Le plan formé par les 2 axes
représentés possède une inertie de 66%. Ceci confère à la figure un poids
suffisant pour évaluer le pouvoir discriminatif des 31 sondes choisis. Les
vecteurs formés par les « variables » souches permettent de constater
que chaque sérovars est bien séparés. Les souches L. innocua sont
bien séparées des L. monocytogenes. Au sein des souches L. monocytogenes,
les sérovars sont séparés et suivent la loi de division génomique
représenté à la Figure 2. En ce qui concerne les sondes, il est intéressant de
remarquer que la sonde lmo0066 participe de manière importante aux sous
groupage des L.innocua 6a. Ceci corrèle bien avec la comparaison des
génomes illustrés sur la Figure
11avec la macro « subtest ».
Dans un deuxième temps les gènes importants donnant des
informations cruciales seront apportés comme par exemple les gènes de virulence
(qui ne sont pas forcement discriminant) [6]. Pour cela, l’apport de ces gènes sera conforté par les
résultats obtenus auparavant avec la puce 1ère génération [7].
Enfin, dans un troisième temps, Pour être plus
discriminatoire, il est nécessaire de diversifier la puce par l’intégration de
gènes spécifiques supplémentaires, issus des séquences de d’autres génomes de
souches de L. monocytogenes mais également par l’intégration de
gènes phagiques, en utilisant des banques de séquences.
Ø
Les autres génomes séquencés de Listeria,
analysées également dans l’unité, proviennent de l’institut de recherche
génomique TIGR. Ces deux séquences proviennent de deux souches de L. monocytogenes
de sérovars 1/2a et 4b. Ces génomes 1/2a et 4b de TIGR ont été comparés par
Blast avec les séquences du génome de L. monocytogenes EGDe, L. innocua
(CLIP11262) et L .monocytogenes 4b (CLIP80459). Les séquences qui
ont une homologie inférieure à 70% seront retenues pour ne sélectionner que les
gènes spécifiques.
Ø
Intégration des gènes
phagiques : ils sont une source à la variabilité des génomes de Listeria.
En effet, un pulsotype différent peut résulter de l’intégration d’un phage.
L’objectif est que le nombre de gènes sélectionnés doit être
le plus représentatif de Listeria et surtout de l’espèce L. monocytogenes,
car c’est celle qui est responsable de la quasi-totalité des cas de listériose.
La puce finale de la 3eme génération (puce 3G) est composée de 317 gènes. Le Tableau 9 présente le nombre des gènes sélectionnés qui ont été
spottées sur la puce 3 G en fonction des souches matrices.
En outre, 165 gènes de la puce sont présents dans le génome
de EGDe, dont 59 lui sont spécifiques. 155 amplicons seront réalisés à partir
de l’ADNg de EGDe. Pour les sondes dérivées des souches de Listeria
autres que EGDe, les amplicons de chaque gène ont été réalisés à partir de 35
paires d’amorce dessinée à partir de la séquence de L. innocua CLIP11262,
61 paires à partir de la séquence de L. monocytogenes CLIP80459 (lm4b),
16 paires L. monocytogenes F2365 (lm4b-T) et 48 paires à partir de la
séquence de L. monocytogenes F6854 du sérovar 1/2a (tg1/2a) et une paire
d’amorce à partir de la séquence de L. monocytogenes CLIP90602 du
sérovar 1/2b (lm12b).
Tableau 9 : Nombre de gènes choisis en fonction de
leur spécificité, de leur présence ou absence par rapport aux souches matrices.
|
Gènes
|
Présent
|
Non présent
|
Spécifique
|
amplicons
|
|
EGDe (lmo)
|
165
|
152
|
59
|
155
|
|
lm4b
|
157
|
160
|
11
|
61
|
|
lin
|
111
|
206
|
23
|
35
|
|
lm4b-T
|
148
|
169
|
13
|
16
|
|
Tg12a
|
nd
|
nd
|
48
|
48
|
|
lm1/2b
|
nd
|
nd
|
1
|
1
|
Légende : lmo pour
les gènes de EGDe ; lm4b pour les gènes de L. monocytogenes 4b ;
lin pour L. innocua 6a ; lm4b-T pour L. monocytogenes 4b
séquencés par TIGR ; tg12a pour les gènes de L. monocytogenes 1/2a
séquencés par TIGR ; lm12b pour les gènes de L. monocytogenes 1/2b.
Les gènes sélectionnés important pour la virulence ou la
différenciation des sérovars de L. monocytogenes ont été choisies
sur la base des résultats de la première membrane (cf. I.A.3) De plus 25 gènes d’origine phagique de Listeria
qui faisait défauts dans les anciennes puces ont été ajouté.
Une étude de l’influence de la concentration de départ
d’ADN génomique sur la PCR a été réalisée pour déterminer la concentration
minimale et nécessaire pour une amplification PCR importante. Le but de cette
étude est d’utiliser le minimum d’ADN génomique pour minimiser le bruit de fond
dû à l’hybridation avec cet ADN.
D’après la Figure
14, la réaction de PCR est variable selon les gènes à
amplifier. Sur cet exemple, les dilutions ont été faites à partir d’un aliquote
d’ADNg de L. innocua de concentration 367µg/ml. Le puit 3 et le
puit 11 semble donner une amplification nécessaire et suffisante soit une
concentration de 3,6ng/µL. Pour effectuer les PCR en masse, il est nécessaire
de trouver une concentration consensus pour tous les ADNg et pour tous les
gènes à amplifier.
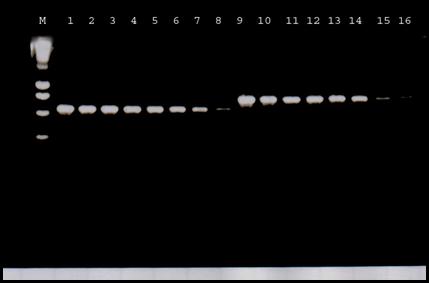
Figure 14 : Photo de migration sur gel en
électrophorèse pour une étude de l’effet de la dilution sur l’amplification par
PCR de deux gènes de L. innocua. M= marqueur de poids moléculaire
Smart Ladder.
|
Tableau des
dilutions
|
10-2
|
2.10-3
|
10-3
|
2.10-4
|
10-4
|
2.10-5
|
10-5
|
2.10-6
|
|
N° Puits
|
Lin0060
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
|
Lin0064
|
9
|
10
|
11
|
12
|
13
|
14
|
15
|
16
|
Sur l’ensemble des
tests qui ont été effectué, l’amplification PCR est bonne lorsque que la
concentration de l’ADNg à amplifier est au moins de 3,5 ng/µL, la concentration
qui a donc été choisie pour l’amplification en masse.
Lors de l’amplification par PCR en masse, il intéressant de
noter que l’amplification varie en quantité et en qualité selon les gènes. Il
semble alors intéressant d’étudier à travers l’élaboration d’un prototype
l’influence de l’amplification PCR sur l’intensité du signal et la formation
systématique de faux négatif ou faux positif lors de l’analyse. J’ai construit
un prototype va être construit avec 192 gènes sélectionnés de EGDe et L. monocytogenes
4b.
|
Figure
15 : Photo
sous UV d’un gel de migration en
electrophorèse de 96 amplicons de gènes lmo
|
|
Qualité d’amplification
|
illustrations
|
résultats
|
|
Peu ou pas d’amplification
|

|
génération de faux négatif
|
|
Présence de double bande
|

|
génération de faux positif systématique
|
|
Bonne amplification
|

|
Bonne sonde
|
Ces différents produits PCR
ont été utilisés pour construire une puce prototype qui a été hybridée avec
les ADNg des souches EGDe et 4b pour déduire l’influence de produits PCR sur
l’intensité des spots. Il résulte que la qualité du signal dépend de la qualité
du produit PCR.
|
Pour l’étape d’hybridation, il a été nécessaire d’étudier
l’influence de température d’hybridation en réalisant parallèlement une
hybridation à 60°C et à 65°C avec le prototype.
|

Figure 16 : Intensités normalisées classées par ordre croissant
Hybridation de la souches EGDe sur le prototype aux température de 60°C et
65°C
|
|
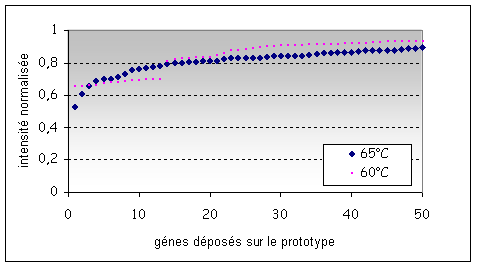
Figure 17 : Intensités normalisées situées dans le bruit de fond
|
La température
influence la stringence. Plus la température est basse plus la stringence est
basse. Une faible stringence peut être intéressante pour les gènes présentant
un polymorphisme élevé (par exemple un gène de L. innocua par
rapport à une sondes d’ADNg de L. monocytogenes). Cependant une
faible stringence augmente le bruit de fond (Voir Figure 17). Malgré une température élevée dans les plus hautes
intensité, la température de 60°C a été choisi pour baisser la stringence et
permettre aux ADN des autres espèces de Listeria de s’hybrider sur la puce (Voir
Figure 16).
La membrane peut être une
source de bruit de fond technique. Quatre lots de membrane ont été préparées avec
les 317 gènes sélectionnés pour la puce 3 G puis comparées par hybridation
de l’ADNg de EGDe. La différence entre ces quatre types de membrane (Performa2 ;
Millipore, NC2015 et Amersham) se fait sur le bruit de fond et sur la zone
d’ambiguïté.
|

Figure 18 : Valeur ratio classé par ordre croissant
|
La Figure
18 présente les valeurs ratio d’une hybridation de la
souche EGDe sur la puce 3G avec les quatre lots de membrane. Les valeurs
ratio diffèrent peu d’un lot de membrane à l’autre. Il faut s’appuyer sur le
bruit de fond et la zone d’ambiguïté pour designer la meilleure membrane de
nylon.
|
|

Figure 19 : Valeurs ratio situé dans le bruit de
fond
|
La Figure
19 illustre les 50 premiers spots qui donnent des
signaux dans le bruit de fond. Il semble que la membrane d’Amersham ait un
bruit de fond moins élevé.
La Figure
20 montre la zone d’ambiguïté où se situe le seuil de
coupure.
|
|
  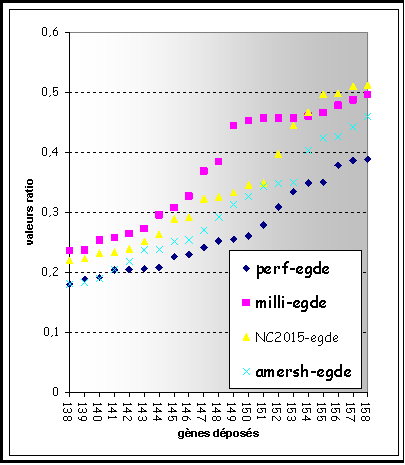
Figure 20 : Seuil de coupure et zone d’ambiguïté
|
Théoriquement, L’ADNg de EGDe n’hybride pas sur les 148
premières sondes (Voir Tableau
9). Le seuil de coupure étant placé à une valeur
ratio de 0,3, le nombre de faux négatif et de faux positif peut donc être
dénombré. Dans cette étude, la membrane de nylon d’Amersham semble être la
plus juste car elle ne forme ni de faux négatif ni de faux positif.
|
Cependant, il faut émettre des réserves à ces observations
car finalement le seuil de coupure fixé à 0,3 (définit expérimentalement avec
les puces précédentes) peut être maintenant différent avec cette nouvelle puce.
Tableau 10 : Proportion de valeur ratio se situant
dans la zone d'ambiguïté définit entre 0,2 et 0,4
|
Membrane
|
performa 2
|
millipore
|
NC2015
|
Amersham
|
|
% de signaux ambigus*
|
5,21%
|
6,78%
|
5,84%
|
4,42%
|
*hybridation avec ADNg de EGDe
D’après le Tableau
10, la proportion de signaux ambigus est plus faible en
utilisant la membrane Amersham. Cependant, la différence de proportions entre
les membranes est faible.
Remarque : Dans tous les cas, la proportion de signaux
ambigus est bien inférieure avec celle observé pour l’hybridation de l’ADNg de
EGDe avec la puce de 2eme génération (12,7%).
Afin que la puce 3G soit
utilisée à travers tous les laboratoires intéressés par cette méthode de
typage, notamment l’AFSSA de Ploufragan, il est nécessaire de comparer les
méthodes d’extraction d’ADNg.
En partenariat avec l’AFSSA, deux kits d’extractions d’ADN
génomique, Wizard et Quiagen, ont été testés. Les experts de l’AFSSA de Ploufragan
en épidémiologie moléculaire ont réalisé quatre paires d’extraction sur des
souches environnementales de L. monocytogenes.
La comparaison des signaux d’intensité entre deux expériences
montre qu’il y a peu de différences entre les kits d’extraction. Cependant,
cette expérience nous a montré l’importance de la concentration d’ADN, si
l’extraction d’ADNg est insuffisante, les signaux se révèlent plus faible.
Cette comparaison montre que la technique de puce à ADN peut s’adapter à différents
types d’extraction d’ADNg.
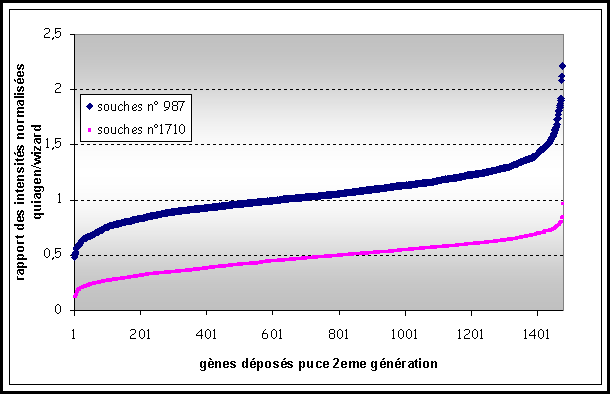
Figure 21 : Rapport des intensités normalisées
quiagen/wizard classés par ordre croissant lors de l’hybridation sur la puce de
2eme génération
La Figure
21 illustre deux paires d’hybridation avec les ADNg de
souches n°987 et n°1710 provenant de l’AFSSA de Ploufragan. Dans le cas de la
souche n°987, les extractions montrent peu de différences car les rapports ne
sont pas supérieurs à un facteur 2 (à 4 exceptions près). En revanche, pour les
souches n°1710, l’extraction d’ADNg avait été mal réalisé avec le kit Quiagen.
Ceci explique que les intensités sont bien supérieures avec le kit Wizard par
rapport au kit Quiagen.
L’hybridation des souches de
référence a permis de construire l’arbre de base de regroupement hiérarchique
sur J-express. Les souches hybridées avec la puce 3 G forment des sous
groupes qui corrèlent avec ceux formés par les anciennes puces. En effet, les
espèces sont séparées et les diffèrent sérovars de L. monocytogenes
sont regroupés entre eux. Les deux divisons génomiques illustré à la Figure 2 sont également présentes avec les sérovars 1/2b, 3b,
4b, 4d d’une part et les sérovars 1/2c, 1/2a, 3a et 3c d’autre part (Voir
Annexe).
Pour tester la faculté de la puce 3 G à séparer les
sérovars et à intégrer des nouvelles souches autre que les références dans la
base de données, 4 souches provenant de Finlande (N° 3, 5, 9, 12) ont été
hybridées. Auparavant, J’avais analysé les 20 souches responsables d’épidémie
en Finlande par hybridation ADN/ADN sur la puce de 2eme génération. Le sérotype
de ces souches (numérotées de 1 à 20) sont connues. De la même manière que la
puce de 2eme génération, les quatre souches se sont regroupés avec les autres
souches de leur sérovars respectifs : la n°3 et 5 avec les 1/2c, la n°9
avec les 1/2a et la n°12 avec les 1/2b.
Afin d’évaluer l’influence de l’apport de sondes provenant
du génome de TIGR 1/2a, trois souches de L. monocytogenes de
sérovars 1/2a isolé en France ont été hybridées. Les souches ont été regroupées
avec les autres souches du sérovars 1/2a. La souche de portage L. monocytogenes
CLIP93666 s’est regroupée avec la souche TIGR 1/2a tandis que les souches
environnementales (CLIP93149 et CLIP93655) forme un autre sous groupe avec EGDe
(Voir Figure 22).
Figure 22 : Formation
du sous groupe de souches de sérovars 1/2a, 1/2c, 3a et 3c
Le CDC d’Atlanta avait envoyé 11 souches au laboratoire
numéroté de 1 a 11 pour une étude en aveugle. Ces souches correspondent à 2
épidémies (1 à 8 d’une part et 9 à 11 d’autre part). Les données
épidémiologiques montraient que ces souches correspondaient à la même épidémie
respectivement. Elles différaient pourtant très légèrement lors de l’analyse en
PFGE. L’objectif de cette étude était de tester la valeur de typage des puces
sur cette question particulière.
Avec la puce de 2eme génération, les souches, 9 et 10, forment
un groupe de souches à part par rapport aux souches n°1 à 8. Les souches 1 et 2
sont très proches et ne différent que d’un phage. Les souches 6 et 8 sont très
proche voire identique. La souche 5 ne diffère des souches 8 et 6 que par
l’absence d’un phage. Cependant selon le CDC, la souche 5 est différente des
souches 8 et 6. Les souches 3, 4 et 7 forment un groupe. La souche 3 diffère
des souches 4 et 7 par la présence d’un phage. La souche 7 diffère des souches
3 et 4 par l’absence d’un autre phage.
Avec la nouvelle puce, le regroupement sur J-Express permet
aussi de séparé les souches 9 et 10 des souches 1 à 8 (voir Figure 23).
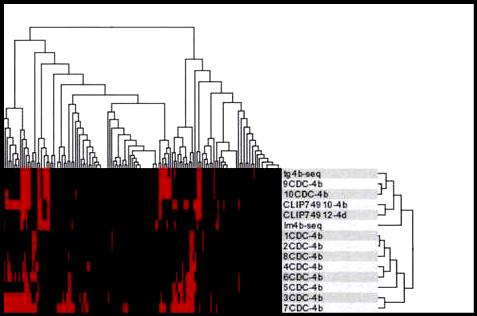
Figure 23 : Regroupement des souches 4b et des
souches du CDC hybridées sur la puce 3 avec J-Express
Cependant avec J-Express la souche n°4 ne semble pas
clustériser comme la puce de 2eme génération. En effet sur J-Express, elle
clustérise avec la souches n°6.
Figure
24 : Regroupement des souches hybridées avec
la puce 3G avec X-strain. Les dénominations des souches sont accompagnées de
leurs sérovars
En revanche la simulation avec X-strain est plus conforme
avec les observations réalisées avec la puce de 2eme génération. Selon le CDC,
la souche 5 qui ne devait pas être aussi proche des souches 6 et 8 se retrouve dans
les deux logiciels légèrement séparés. Comme précédemment les souches 9 et 10
clustérisent dans un autre sous-groupe. La souche 11 n’a pas été analysée avec
la puce de deuxième génération.
Une analyse plus fine des résultats montre que la puce
3 G identifie mieux les différences entre ces souches. Cette amélioration
de sensibilité est due à l’ajout des sondes phagiques et de sondes provenant de
la souche TIGR 1/2a. Par ailleurs, les sondes qui ont été enlevées dans la puce
3 G comparé à la puce de 2eme génération n’ont pas d’impact sur la valeur
discriminante de la puce.
La réalisation d’une puce in silico de 31 sondes a permis de
partir sur une base minimum pour discriminer les espèces et les sérovars. La
séparation et le regroupement des souches suivent bien le principe de lignage défini
par l’analyse en champ pulsé. L’ajout progressif de sondes informatives permet
d’améliorer le pouvoir discriminant au niveau de celui de la puce de 2eme
génération avec un nombre de sondes divisé par 2,5. L’apport ensuite de
nouveaux gènes absents de EGDe provenant d’une autre souche de sérovars 1/2a a
permis par regroupement hiérarchique de former un sous groupe supplémentaire
dans les souches de sérovars 1/2a. En reprenant l’exemple des souches du CDC,
il se peut que l’intégration plus importante des gènes de phage ait permis une
séparation plus nette de la souche n°5 par rapport aux souches n° 6 et 8.
Cependant la constitution d’une base de données plus importante après
hybridation d’un grand nombre de souches par le CNR Listeria devrait par
la suite appuyer ce constat.
L’amplification PCR est l’étape cruciale pour la réalisation
de la puce. Si la concentration intervient dans la qualité des taches, la
qualité de l’amplification doit être bonne afin d’éviter la génération
systématique de faux positif ou de faux négatif.
La température d’hybridation a également une influence sur
les intensités des signaux. Une température de 65°C peut être une bonne
solution pour constituer une base unique de L. monocytogenes
phylogénétiquement proche avec un bruit de fond plus faible. En revanche, une
température de 60°C est plus appropriée pour hybridées d’autre espèce comme L. innocua,
ou même pour assurer une bonne hybridation des souches des 2 lignages de L. monocytogenes.
L’étude sur les différents lots de membrane a permis de
montrer qu’ils influencent aussi la qualité des signaux. Le bruit de fond est
variable mais on ne peut conclure sur la zone d’ambiguïté. Les membranes de
nylon sont sans doute le matériel qui influence le moins l’intensité des
signaux.
L’utilisation de la puce à ADN est flexible. En effet, elle
peut s’adapter à plusieurs kits d’extraction. L’essentiel est que la
concentration d’ADNg extraite soit suffisante pour le marquage sinon
l’intensité des signaux est faible ainsi que la rapport signal/ bruit.
En séparant les espèces, les sérovars et en étant conforme
avec les divisions génomiques, la puce 3G a montré sa fiabilité. Elle est au
moins aussi discriminantes que la puce de 2eme génération. Cependant la
constitution d’une base donnée de plusieurs centaines de souches pourrait
conforter ce potentiel de discrimination.
La puce 3G permet d’intégrer dans sa base d’autres souches
qui se regroupe avec leur sérovars respectif. En revanche pour les souches très
proches phylogénétiquement, l’interprétation peut être différentes selon les
logiciels utilisés. L’intégration de gènes de phage peut permettre de séparer
au sein d’un sous groupe des souches très proche. C’est pour cette raison en
partie que la souche n°5 du CDC a pu être séparée des souches 6 et 8.
Le nombre de sondes dans cette nouvelle puce a été réduit
par un facteur deux avec 317 gènes sélectionnées. De plus, elle garde le même
pouvoir discriminatoire que l’ancienne et se diversifie avec l’intégration de
nouveaux gènes. A ce propos même si la proportion de sondes de EGDe reste très
importante sur la puce, elle n’en demeure par moins représentatif puisque
certaines sondes, comme lmo0066, sont contenues dans le génome de L. innocua
et participe à la séparation des souches.
En comparant l’hybridation de EGDe entre la puce de 2eme
génération et la puce 3G, la proportion de signaux ambiguës est réduite de prés
de 58,7%. Cependant, il serait nécessaire d’hybrider plusieurs centaines de
souches pour estimer plus significativement la baisse réelle de signaux
ambigus.
Pour éviter le problème de cette étape de binarisation, l’utilisation
de « X-strains » semble être un bon complément de J-express. En
effet, cette méthode de classification corrèle bien avec J-Express et semble
être encore plus juste pour les souches très proches comme celle du CDC.
Afin d’apprécier et de gérer le risque lié à une
contamination à Listeria, la recherche scientifique a fait évoluer les
méthodes de typages de plus en plus discriminantes. En entrant dans l’ère de la
post-génomique, il est possible d’aller plus loin grâce à l’émergence des puces
à ADN. Les puces à ADN permettent d’obtenir des informations fonctionnelles et
d’analyser l’évolution de la diversité au sein d’une espèce.
Comme toute nouvelle méthode, le développement d’une puce à
ADN pour le typage de Listéria a nécessité plusieurs mises au point. Celle-ci
requiert un intérêt particulier aux valeurs ambiguës, source d’erreur dans la
différenciation des souches. L’effort employé pour réduire les ambiguïtés a
nécessité dans un premier temps une sélection discriminante de bonnes sondes
mais aussi une étude importante sur l’influence de toutes les étapes de
fabrication de la puce mais aussi lors de son utilisation.
Cette troisième génération de puce à ADN pour le typage de Listeria
est au moins aussi discriminantes que les précédentes puces mais elle donne une
part plus importante aux gènes de phage qui permettent de différencier les
souches épidémiques très proches.
Le but final de cette puce est d’être intégré dans un
processus de routine destiné au laboratoire de typage de Listéria. La réduction
du nombre de gènes pourrait contribuer aussi à la réduction de sa taille
physique afin de l’intégrer dans un appareil automatisable. Cependant,
l’amélioration majeure serait d’utiliser un procédé de révélation autre que la
radioactivité. Celle-ci ne pouvant pas être utilisé dans tous les laboratoires,
il faudra réadapter son protocole d’utilisation. Une étude de faisabilité de
l’utilisation de la chemiluminescence a donné des résultats très encourageant
pour cette alternative non radioactive
[1] AFSSA, Rapport de la commission d’étude des risques liés à Listeria
monocytogenes (2000)
[2] R Brosch, J Chen, and J B Luchansky. Pulsed-field
fingerprinting of listeriae: identification of genomic divisions for Listeria
monocytogenes and their correlation with sérovar (1994) 60(7):2584-92
[3] Buchrieser C, Runiok,
et al., Comparaison of the genome sequences of Listeria monocytogenes
and Listeria innocua: clues for evolution and pathogenicity Immunology
and medical microbiology (2003) 35: 207-213
[4] Cai S, Kabuki DY, Kuaye AY, Cargioli TG, Chung MS, Nielsen R, Wiedmann M Rational design of DNA sequence-based strategies for subtyping Listeria
monocytogenes. J Clin Microbiol. (2002), 40:3319-25.
[5] Croizé J. Listériose : un problème de santé ancien ou actuel ? La
Revue de Médecine Interne. Février 2000, Vol. 21, n°2, Pages 143-146.
[6] Doumith M,
Buchrieser C, Glaser P, Jacquet C, Martin P.Differentiation of the major Listeria
monocytogenes serovars by multiplex PCR.J Clin Microbiol. (2004)
;42:3819-22
[7] Doumith M, Cazalet C, Simoes N, Frangeul L, Jacquet C, Kunst F, Martin P, Cossart P, Glaser P, Buchrieser C., New aspects regarding evolution
and virulence of Listeria monocytogenes revealed by comparative
genomics and DNA arrays. Infect Immun. (2004), 72 :1072-83
[8] Dussurget O., Pizarro-Cerda J., et al., Molecular determinants of Listeria
monocytogenes Virulence, Annu. Rev. Microbiol (2004) 58 :587-610
[9] Dysvik B, Jonassen I.
J-Express: exploring gene expression data using Java. Bioinformatics. (2001);
17(4):369-70
[10]
Evans M.R., Swaminithan B. et al, Genetic
markers unique to Listeria monocytogenes Serotype 4b
differentiate epidemic clone II (hot dog outbreak strains) from other lineages,
applied and environemental microbiology (2004) 70: 2383-2390
[11]
Grimont, F. and Grimont, P.A.D. Ribosomal
ribonucleic acid gene restrictions patterns as potential taxonomic tools. Ann.
Inst. Pasteur/Microbiology (1986). 137B: 165-175.
[12]
Glaser P, Frangeul L, Buchrieser C, Rusniok C, Comparative genomics of Listeria
species. Science. (2001) 294 :849-52.
[13]
Glaser P., Les puces à ADN
vont-elles révolutionner l’identification des bactéries? Médecine/Sciences
(2005) ; 21 :539-544
[14]
Goulet V., Jacquet Ch, et al.
La surveillance de la Listériose humaine en France en 1999. Bulletin
épidémiologique hebdomadaire [en ligne]. Août 2001, n°34
[15]
Herd M, Kocks
C., Gene fragments distinguishing an epidemic-associated strain from a
virulent prototype strain of Listeria monocytogenes belong to a
distinct functional subset of genes and partially cross-hybridize with other Listeria
species.Infect Immun. (2001), 69:3972-3979.
[16]
HuyenL., Kathariou S, Restriction fragment
length polymorphisms detected with novel DNA probes differentiate among diverse
lineages of serogroup 4 Listeria monocytogenes andidentify four
distinc lineages in serotype 4b, (2002) 68: 59-64
[17]
Jacquet C., Catimel B., et al.
Investigations Related to the Epidemic Strain
Involved in the French Listeriosis Outbreak in 1992, (1995), 61 : 2242–2246
[18]
Lewis M. Graves, Susan B. Hunter,
Microbiological Aspects of the Investigation That Traced the 1998 Outbreak of
Listeriosis in the United States to Contaminated Hot Dogs and Establishment of
Molecular Subtyping-Based Surveillance for Listeria monocytogenes
in the PulseNet Network, Journal of clinical microbiology, (2005), 2350–2355
[19]
Loesner M., Inman R.B., et al, Complete
nucleotide sequence, molecular analysis and genome qtructure of bacteriophage
A118 of Listeria monocytogenes: implication for phage evolution,
Molecular microbiology (2000) 35:324-340
[20]
Lucchini S., Thompson A., et al;, Microarray
for microbiologists, Microbiology (2001) 147: 1410-1414
[21]
Martin P., CNR des Listeria.
Rapport d’activité de l’unité Listeria pour l’année 2001. [en ligne] disponible
sur : http://www.pasteur.fr/recherche/RAR/RAR2001/Listeria.html.
[22]
Nelson KE, Fouts DE, et al,.Whole
genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen
Listeria monocytogenes reveal new insights into the core genome components of
this species.Nucleic Acids Res. (2004) 32 : 2386-95.
[23]
Olier M, Garmyn D, Rousseaux S, Lemaitre JP, Piveteau P, Guzzo J.Truncated internalin A and asymptomatic Listeria monocytogenes
carriage: in vivo investigation by allelic exchange. Infect Immun. (2005), 73
:644-8.
[24]
Rusniok C, Jones LM, Duperrier S, Tap J, Kunst
F, Buchrieser C et Glaser P. X-strain- a method to groupe bacterial strains
according to DNA DNA-array hybridization data. Poster Institut Pasteur (2005).
[25]
Ward TJ, Gorski L, Borucki MK, Mandrell RE, Hutchins J, Pupedis K., Intraspecific phylogeny and lineage group identification based on the
prfA virulence gene groupe of Listeria monocytogenes. J
Bacteriol. (2004) 15:4994-5002.
[26]
Zhang W, Jayarao BM, Knabel SJ., Multi-virulence-locus sequence
typing of Listeria monocytogenes.Appl Environ Microbiol. (2004),
70 :913-20.
[27]
Wiedmann M, James B.L., Ribotype diversity of
listeria monocytogenes strains associated with outbreaks of listeriosis in
ruminants Journal of clinical microbiology,(1996) , 34:1086–1090.
[28]
Zhang C., Zhang M, et al., Genome
Diversification in philogenic lineages I and II of Listeria monocytogenes:
Identification of segments unique to lineage II populations, Journal of
bacteriology (2003) 185: 5573-5584
[29]
Zimmer M., Sattelberger E, et al., Genome and
proteome of Listreria monocytogenes phage PSA: an unusual case for
programmed +1 translational frameshifting in structural protein synthesis,
Molecular microbiology (2003) 50:303-317
Figure
1 : Protéine de surface liés par covalence avec la paroi cellulaire contenant
un motif LPxTG.. 3
Figure
2 : Représentation des différentes lignées à partir d’un PFGE numérisé de 62
souches représentatives de L. monocytogenes [2] 8
Figure
3 : Analyse d’acides nucléiques par puce à ADN.[13] 12
Figure
4 : Scan de la membrane portant 739 sondes en duplicata hybridée par l’ADNg de L. monocytogenes
marquée radioactivement au 33P 17
Figure
5 : Quantification des signaux avec Array-vision. 17
Figure
6 : Intensité brute classées par ordre croissant resultant de
l'hybridation d'une souche de L. monocytogenes provenant de
Helsinki 18
Figure
7 : Rapports classés par ordre croissant des intensités normalisées entre
le signal et la référence en fonction des gènes (valeurs ratio), ADNg d’une
souche L. monocytogenes en provenance de Helsinki hybridées sur la puce de
2eme génération 19
Figure
8 : Interface du logiciel J-Express. 21
Figure
9 : Interface du logiciel de pilotage « Gridding » du Qpix de Genetix. 29
Figure
10 : Organigramme décisionnel "if then else". 36
Figure
11 : Illustration de l'utilisation de la macro « subtest » dans la
cas d'une recherche rapide de gènes lin dans le génome de EGDe 38
Figure
12 : Présentation des étapes de selection des gènes. 39
Figure
13 : Analyse en Composantes Principales d’une simulation « in
silico », d’hybridation de 10 souches de référence avec les 31 sondes
choisis dans la Figure 12. 40
Figure
14 : Photo de migration sur gel en électrophorèse pour une étude de l’effet de
la dilution sur l’amplification par PCR de deux gènes de L. innocua.
M= marqueur de poids moléculaire Smart Ladder. 43
Figure
15 : Photo sous UV d’un gel de migration
en electrophorèse de 96 amplicons de gènes lmo. 44
Figure
16 : Intensités normalisées classées par ordre croissant Hybridation de la
souches EGDe sur le prototype aux température de 60°C et 65°C.. 44
Figure
17 : Intensités normalisées situées dans le bruit de fond. 45
Figure
18 : Valeur ratio classé par ordre croissant 46
Figure
19 : Valeurs ratio situé dans le bruit de fond. 46
Figure
20 : Seuil de coupure et zone d’ambiguïté. 47
Figure
21 : Rapport des intensités normalisées quiagen/wizard classés par ordre
croissant lors de l’hybridation sur la puce de 2eme génération. 48
Figure
22 : Formation du sous groupe de souches de sérovars 1/2a, 1/2c, 3a et 3c. 49
Figure
23 : Regroupement des souches 4b et des souches du CDC hybridées sur la puce 3
avec J-Express. 50
Figure
24 : Regroupement des souches hybridées avec la puce 3G avec X-strain. Les
dénominations des souches sont accompagnées de leurs sérovars. 51
Tableau
1: Sérotypes au sein du genre Listeria. 6
Tableau
2 : Liste des difficultés rencontrées. 22
Tableau
3 : Souches matrices utilisées pour le développement de la nouvelle puce. 25
Tableau
4 : Descriptions des souches à hybrider sur le la puce 3G.. 26
Tableau
5 : Mélange réactionel pour la PCR et programme utilisé pour le thermocycleur
(GeneAmp) 28
Tableau
6 : Paramètres de pilotage du Qpix pour le dépôt des sondes sur la macro-array. 30
Tableau
7 : Solution d’hybridation pour une membrane. 32
Tableau
8 : Macro sub test() applicable sur les logiciels Office Mac OS. 37
Tableau
9 : Nombre de gènes choisis en fonction de leur spécificité, de leur présence
ou absence par rapport aux souches matrices. 42
Tableau
10 : Proportion de valeur ratio se situant dans la zone d'ambiguïté définit
entre 0,2 et 0,4. 47
