Structure génétique de Mycobacterium prototuberculosis et
origine de M. tuberculosis.
Sommaire
I. Introduction................................................................................................................................................................................. 3
A. Les bacilles de la tuberculose............................................................................................................................................ 3
1. Caractéristiques et physiologie.............................................................................................................................................. 3
2. Pathogénicité........................................................................................................................................................................... 3
3. Biodiversité, succès évolutif et
pharmaco-résistance.......................................................................................................... 4
B. Le complexe d’espèce de Mycobacterium tuberculosis (MTBC)............................................................................... 4
1. Taxonomie et écologie............................................................................................................................................................ 5
2. Du point de vue génomique................................................................................................................................................... 6
3. Le nouveau scénario évolutif du
MTBC................................................................................................................................ 7
4. Polymorphisme et transferts
horizontaux : les origines du MTBC.................................................................................... 8
5. La co-évolution hôte pathogène........................................................................................................................................... 9
II. Matériels et méthodes......................................................................................................................................................... 11
A. Souches bactériennes........................................................................................................................................................... 11
B. Sélection des locus pour le séquençage...................................................................................................................... 12
C. Amorces utilisées................................................................................................................................................................... 13
D. Amplification des locus par PCR..................................................................................................................................... 14
E. Séquençage des produits PCR............................................................................................................................................ 15
III. Résultats et interprétations........................................................................................................................................... 17
Résumé
Mycobacterium tuberculosis, l’agent responsable de la tuberculose, est un
pathogène mortel infectant un tiers de la population mondiale. Au sein du genre
Mycobacterium, ces pathogènes
opportunistes obligatoires sont très liés et partage des facteurs déterminant
de virulence en commun. Les mutations, plus que les transferts horizontaux de
gène, ont conduit l’évolution récente de Mycobacterium
tuberculosis et son adaptation à l’hôte humain. La diversité génétique
limitée des souches contemporaines suggèrent que M. tuberculosis est issu d’une récente expansion clonale. La
population de M. tuberculosis est
très fortement structurée géographiquement. L’existence d’un clone dominant au
sein des différentes populations humaines soulève la possibilité d’une co-évolution hôte-pathogene. Les
sous-espèces du complexe de M.
tuberculosis, avec leur diversité d’hôtes, pourraient être issu du progéniteur du pathogène humain.
En 1882, le médecin
allemand Robert Koch réussi à isoler et à cultiver le bacille responsable de la
tuberculose, Mycobacterium tuberculosis
sera aussi appelé par la suite bacille de Koch. La tuberculose est une des
maladies les plus anciennes et un problème majeur à l’échelle mondiale. Selon
l’Organisation Mondiale de la Santé, près d’un tiers de la population est
affecté avec 1,7 millions de décès en 2004. Par ailleurs, une infection due à
un bacille tuberculeux a lieu chaque seconde. C’est en Asie du Sud-Est et en Afrique subsaharienne, que la tuberculose
sévit le plus fortement. Ceci est dû au problème de mal nutrition et au VIH qui
permet une progression plus rapide de la maladie. [18]
Les bacilles tuberculeux
sont caractérisés par une croissance lente, une enveloppe cellulaire complexe,
une pathogénicité intracellulaire et une homogénéité
génétique. Leur temps de génération est de 24 heures. Les mycobactéries
font partie du genre Mycobacteriaceae
dans le sous-ordre Corynebacteriaceae,
ordre des Actinomycetales.
Parmi les mycobactéries, il faut distinguer les bacilles causant la tuberculose
comme M. tuberculosis de M. leprae,
agent de la lèpre, mais également de M. avium, M. marinum M. kansasii et M. xenopi responsable de mycobactérioses.
Les mycobactéries ont une enveloppe cellulaire rugueuse Gram positif avec des
peptidoglycanes riches en lipide "unusuelle".
Ceci représente 20 à 45% de l’ensemble de la bactérie, ce qui rend la bactérie
peu perméable aux éléments hydrophiles. Parmi ces lipides, l’acide mycolique joue un rôle important dans l’acido-alcoolo-resistance.[3]
M. tuberculosis ne produit pas de toxine et infecte les
macrophages de l’hôte. Ce n’est que dans la dernière décennie que les gènes
liées à la virulence ont pu être mis en relief. Ces gènes ont pu être reparti
en plusieurs catégories comme la structure de l’enveloppe cellulaire, la
synthèse de lipide, de gènes de résistance au stress, etc.
Il est intéressant de
noter que M. avium
et M. marinum
partage avec M. tuberculosis la
même stratégie d’infection de l’hôte en survivant et se multipliant au sein des
macrophages. Après la phagocytose, ces derniers inhibent l’acidification et la
fusion entre le phagosome et lysosome leur permettant
de prospérer à l’intérieur du phagosome. M. tuberculosis survit dans le
macrophage grâce à un système de détoxification de radicaux libres et de
chélation du fer. Ce système implique les gènes katG
codant pour la catalase peroxydase, le gène furA codant
pour une protéine de captation du fer et sodA codant
pour la superoxyde dismutase.
[11]
D’autre part, M. tuberculosis survie dans le
macrophage grâce à son enveloppe cellulaire. Des voies de biosynthèses
spécifiques sont impliquées dans la formation de cette enveloppe cellulaire
comme celle de l'acide mycolique. La synthèse des
acides mycoliques est souvent liée à des gènes
déterminants dans la virulence comme fbpA, mmaA4 et
LAM. L’acide mycolique est un responsable important
dans le rôle protecteur de l’enveloppe cellulaire chez M. tuberculosis. Plus spécifiquement, les motifs cyclopropanes
permettent d’assurer l’intégrité de l’enveloppe cellulaire et protége le bacille des stress oxydatifs comme ceux qui sont
générés dans un phagosome. Par ailleurs, il
semblerait que le gène pks12, possédant la plus grande phase ouverte de lecture
dans le génome de M. tuberculosis,
est directement impliqué dans la pathogenicité du
bacille car il encoderait deux modules catalysant la formation des acides
saturées tels que les acides mycoliques. [13]
On ne distingue pas assez
les gènes qui sont impliqués dans la virulence même des gènes qui permettent de
s’adapter à l’hôte. Par exemple, l’enveloppe cellulaire des Mycobacterium est très complexe et
permet une protection aux stress environnementaux. Les gènes impliqués dans la
formation de l’enveloppe cellulaire sont très conservés. Ces gènes peuvent être
considéré comme des facteurs déterminants à la virulence car ils ont permis à M. tuberculosis de coloniser de
nouvelles niches écologiques et de contribuer à son succès évolutif du fait de
son expansion clonale sur tous le globe.
Il y a cinquante ans
encore, il n’existait aucun médicament contre la tuberculose. L’effort fourni
dans le domaine médical et de santé publique a permis de comprendre dans les
moindres détails l’épidémiologie, les manifestations cliniques et le traitement
de cette maladie. Cependant aujourd’hui, certaines souches deviennent
résistantes à certain médicament utilisé seul voire à plusieurs
antituberculeux. On parle alors de souches pharmaco-résistantes. Pour
comprendre l’émergence de ces résistances, il est nécessaire d’étudier
l’histoire évolutive et la diversité génétique de ces bacilles tuberculeux et
en particulier celle qui est liée au complexe d’espèce M. tuberculosis par une approche de génétique de population
bactérienne. Ces nouvelles connaissances, notamment dues au progrès en
génomique, devraient contribuer au développement de nouvelles immunothérapies. [18]
Le but de la recherche en
génétique de population bactérienne est de comprendre les relations entre la
diversité génétique, les lignées clonales, et leurs phénotypes comme la
virulence, la transmissibilité, la spécialisation de l’hôte et le succès
évolutif. [8] Dans le cadre des bacilles tuberculeux, il est
nécessaire de s’intéresser en particulier au succès évolutif d’un clone celui
de M. tuberculosis et de son complexe
d’espèce. Toutes les souches de M.
tuberculosis sont définies comme étant un unique clone car elles ont toutes
le même génotype.
Le complexe d’espèce M. tuberculosis (MTBC) constitue un
groupe très compact, et ses membres M. tuberculosis,
M. africanum,
M. bovis, M. pinipedi, M. caprae et M. microti
peuvent être considéré comme des sous- espèces de M. tuberculosis. Cette compacité du MTBC a été établie par
hybridation ADN-ADN (>95%), séquençage de l’ARN ribosomique
16S et des gènes de ménages. Ceci marque une diversification récente du clone M. tuberculosis formant alors ce
complexe clonal. De plus, les variations au sein des éléments d’ADN répétés,
comme les séquences d’insertion IS6110 et les répétitions directes (DR) ont été
trouvées restreintes dans le MTBC. [7]
Néanmoins, la niche
écologique et la pathogénicité des espèces du
complexe du M. tuberculosis varient
énormément. Le réservoir naturel de M. tuberculosis
et de M. africanum
est limité à l’Homme tandis que celui de M. microti est limité aux rongeurs. Par contre, malgré une
préférence pour les bovins, la niche écologique de M. bovis est très large. En effet,
cette espèce provoque des maladies dans un large panel de mammifère domestique
et sauvage, y compris les humains. [17]
L’analyse par séquençage de 26 gènes structurels a montré une faible proportion de substitutions nucléotidiques synonymes dans le complexe d’espèce de M. tuberculosis comparé aux autres bactéries pathogènes. En accord avec les précédentes études éléctrophorétiques, cette faible proportion de mutation neutre dans les gènes structurels (ou de ménage) indique que M. tuberculosis est issu d’une évolution récente avec une expansion clonale globale. [15]

Figure 1: Scénario évolutif de M. tuberculosis selon Streevatsan et al. Le précurseur du complexe de
M. tuberculosis est caractérisé par le codon 463 katG
et le codon 95 de gyrA. Ces deux sites utilisés comme
marqueurs génétiques permettent d’identifier trois groupes de
M. tuberculosis. [15]
L’équipe de Stewart Cole
a réalisé le séquençage en 1997 de la souche de laboratoire de référence H37Rv.
Le génome de Mycobacterium tuberculosis
H37Rv a une taille d'environ 4 Mb dont environ 4000 gènes. M. tuberculosis diffère des autres
génomes bactériens par sa grande proportion de séquences codantes impliquées
dans la production d'enzyme de la lipogenèse et de la lipolyse. Chez M. tuberculosis, les codons
initiateurs de la transcription sont GTG, ATG et ATC. [3]
Le génome de M. tuberculosis est caractérisé par un
haut G+C% de 65% qui reste relativement constant à travers le génome. Ceci
indique la très faible possibilité de transferts horizontaux de gènes alors que
ces derniers peuvent contribuer au succès évolutif en donnant, par exemple, la
capacité d’infecter de nouveaux hôtes. Certaines séquences possèdent un très
haut niveau de G+C% supérieur à 65%. Ces séquences sont appelées PGRS (Polymorphic G+C Rich Sequences) [3]
Le séquençage du génome
de M. tuberculosis a mis en
évidences 2 nouvelles familles de protéine riche en glycine avec des structures
répétées et qui seraient impliquées dans des variations antigéniques. Le génome
est riche en ADN répété et particulièrement en séquences d'insertions mais
aussi l'apparition de nouvelle famille multigénique
et des gènes de ménages dupliqués. Les séquences d'insertions sont insérées
dans des régions intergéniques ou non codante. La
plupart de séquences sont regroupés formant alors des "hot spot"
d'insertion. [3]
10% du génome code pour
des familles de protéine PE et PPE. Les protéines PE sont caractérisées par un
motif Pro-Glu et PPE par un motif Pro-Pro-Glu localisé à proximité du N-
terminal. Ces familles de protéines seraient impliquées dans un rôle immunologique.
[3]
Le premier scénario
évolutif tentait a montré que M. bovis fut le progéniteur de M. tuberculosis. On pensait que M. bovis
s’était adapté à l’homme lors de la domestication des bovins. En effet, Mycobacterium tuberculosis possède un
faible niveau de variation génétique ce qui conduit à penser qu'il y a eu une
expansion d'un clone, il y a 35000 ans seulement.
La plupart des espèces
ont un large spectre de clone distinct avec plus de 1% de variation de
nucléotide synonyme généré par mutation et transferts horizontaux.. Cependant
dans le MTBC, chaque membre a un impact différent mais une très grande homogénéité
avec 0,01% de variation sans trace d'échange génétique. Le MTBC proviendrait
d'un ancêtre commun qui aurait connu un goulot d'étranglement entre 20000 et
35000 ans.[15]
L’analyse basée sur la génomique comparative de la distribution de 20 régions variables résultant d’événement d’insertion et de délétion a été effectuée sur 100 souches de M. tuberculosis. Cette approche a permis de bâtir un nouveau scénario évolutif pour le MTBC où non seulement M. tuberculosis ne proviendrait pas de M. bovis mais que le variant M. canettii, de phenotype lisse, serait antérieur au précurseur de ce complexe clonal. [1]
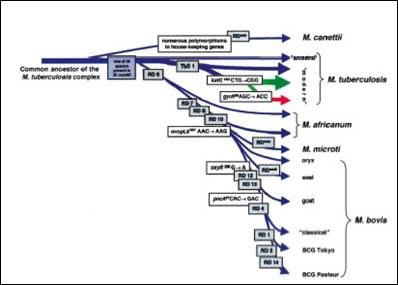
Figure 2: nouveau scénario évolutif proposé par Brosch et al. Ce schéma est basé sur la présence/absence de
région conservée et sur le polymorphisme de 5 gènes sélectionnées. La nouveauté
est de proposer que M. canetii de phénotype
« lisse » est antérieur au précurseur de M. tuberculosis.
Après l’établissement de
ce nouveau scénario évolutif, il était intéressant d’orienter les recherche sur
l’ancêtre commun de ce complexe d’espèce pour comprendre son origine et son
succès évolutif. L'isolement de nouveaux bacilles tuberculeux à Djibouti
provoquant la tuberculose a permis d'apporter de nouvelles connaissances sur
l'histoire évolutive des Mycobacterium et en particulier du MTBC.
Contrairement à M. tuberculosis, ces nouvelles souches
forment des colonies lisses comme M. canetii, le dernier bacille de la tuberculose à avoir
été décrit. [17] La phylogénie réalisée à partir des gènes de ménages (n = 6) permet de
former un groupe compact entre les souches lisses (n = 37) et le MTBC. Par ailleurs,
on n'observe ni de délétion ni d'insertion mais 52 sites polymorphes dont 46
substitutions synonymes. La distance entre les allèles du MTBC est inférieure à
celle des souches lisses. [9]
Le MTBC serait donc un
sous-groupe des souches lisses. Les distances synonymes entre tous les allèles
des bacilles tuberculeux sont similaires à ceux d’une espèce. La plupart des
substitutions non synonymes sont dans les souches lisses. Le rapport Ks/Ka = 33% est proche des valeurs observées des autres
espèces bactériennes supérieure au 1,6% entre les souches de M. tuberculosis. Un rapport Ks/Ka élevé traduit une longue période de changement tandis
qu'un rapport Ks/Ka faible traduit une expansion
récente. MTBC provient donc d'une plus grande et ancienne espèce bactérienne
baptisée du fait de son antériorité M. prototuberculosis.
[9]
L'alignement des gènes de ménage a permis de mettre en évidence une structure de gènes mosaïques notamment dans gyrA et gyrB. ceci montre qu'il y eu des recombinaison intra génique dans les souches lisses. Ces mutations et ses recombinaisons témoignent de l'évolution des bacilles lisses tuberculeux. La concaténation des alignements des gènes de ménage fait apparaître des structures sous forme de patch témoignant d'événement de transferts horizontaux. L’alignement des séquences concaténées de 3, 387 kpb de chaque souches lisses et rugueuses ont permis de mettre en évidence 52 sites polymorphes dont 46 sites synonymes et sans aucune insertion - délétion. Le peu de recombinaison actuelle est peut-être dû au fait qu'il ait eu une perte de la capacité des transferts horizontaux, que les événements de transferts horizontaux soient trop rares ou bien que la niche écologique de ces bacilles soit différente écartant l'opportunité de transferts. [9]

Figure 3 : Split tree réalisée à
partir des 17 séquences concaténées des 6 gènes de ménages. Le MTBC de
phénotype rugueux forme avec les bacilles tuberculeux lisses une nouvelle
espèce baptisée M. prototuberculosis. L’échelle représente la distance de Hamming. [9]
Ces bacilles tuberculeux
lisses auraient une origine de 2,6 à 2,8 millions d’années et ils ont été
isolés sur des patients atteints de la tuberculose. Par conséquent, le dernier
ancêtre commun entre ces souches lisses et ces souches rugueuses pouvait causer
la tuberculose. Ceci confirme que la tuberculose n'est pas récente et beaucoup
plus vieux de plusieurs millions d’années (plus vieille que la fièvre typhoïde
et la malaria). Ces souches lisses ont été isolées en Afrique, là où les premiers
hommes furent sans doute affectés. L'opposition entre la diversité génétique de
ses souches lisses et l'expansion clonale internationalement est à mettre en
parallèle avec l’histoire évolutive de l'homme (l'hypothèse "OUT OF
AFRICA"). Ceci serait dont une preuve de l'existence d'une co-évolution entre l'Homme et l’agent de la tuberculose. [16]
La longue interaction
entre humains et les bacilles tuberculeux est impliquée dès lors dans des
processus de sélections naturelles. Cela va de pair avec les mutations non
synonymes des gènes codant pour les antigènes, des toxines,... Ceci conduit à
réfléchir sur l'immunothérapie et le risque de faire émerger de nouveau variant
en contribuant à l’émergence de souche multi résistance. [9]
Bibliographie
- Brosch R., Gordon SV, et al. (2002) A new evolutonary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A 99 : 3684 – 3689.
- Bulut Y., Michelsen K. S., et al (2005) Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J. Bio. Chem. 280 : 20961-20967
- Cole S.T., Brosch R., et al. (1997) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393 : 537 – 544.
- Fleischmann R. D., Alland D., et al. (2002) Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J. Bacteriology 184 : 5479 – 5490.
- Garnier T., Eiglmeier
K., et al. (2003)
The complete genome sequence et Mycobacterium bovis.
Proc Natl Acad Sci 100 : 7877 – 7882
- Gil R., Silva F. J., et al (2004) Determination of the core of a minimal bacterial gene set. Micr. Mol Biol. 68 : 518 – 537.
- Gordon S. V., Heym B. et al. (1999) New insertion sequences and a novel repeated sequences in the genome of Mycobacterium tuberculosis H37Rv. Microbiology 145 : 881 – 892.
- Gutacker M. M., Smoot J. C., et al (2002) Genome-wide analalysis of synonymous single nucelotide polymorphisms in Mycobacterium tuberculosis complex organism : reslution of genetic relationships among closely related microbial strains. Genetics 162 : 1533 – 1543.
- Gutierrez M. C., Brisse S., et al. (2005) Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. Plos Pathogens 1 : 55 – 61.
- Hughes A. L.., Friedman R., et al. (2002) Genowide pattern of synonymous nucleotide substitution in two complete genomes of Mycobacterium tuberculosis. Emerg. Inf. Dis. 8 : 1342 – 1346.
- Pym A.S., Domenech P., et al. (2001) Regulation of catalase-peroxidase (katG) expression, isonazid sensivity and virulence by furA of Mycobacterium tuberculosis. Mol. Microbiology 40 : 879-889.
- Saint-Joanis B ;, Souchon H., et al. (1999) Use of-directed mutagenesis to probe structure, function and ionazid activation of the catalse/peroxidase, katG, from Mycobacterium tuberculosis. Biochem. J. 338 : 753-760.
- Sirakova T.D. , Dubey V. S. et al (2003) The largest open reading frame in Mycobacterium tuberculosis genome is involved in pathogenesis and Dymycoseryl Phthiocerol Synthesis. Inf and Imm. 71 : 3794 - 3801
- Spratt B. G. (2004) Exploring the concept of clonalty in bacteria. Methods Mol Biol 266 : 323 - 352
- Sreevatsan S, Pan X, Stockbauer KE, et al. (1997) Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A 94: 9869–9874.
- Templeton A. R. (2002) Out of Africa again and again. Nature 416 : 45 – 51.
- Van Soolingen D., Hoogenboezem T. et al. (1997) A novel pathogenic taxoon of the Mycobacterium tuberculsis complex, Canetti : characterization of an excecptional isolate from Africa. Int. J . Syst. Bact. 47 : 1236 -1245.
- World Health Organization. (2006) Global tuberculosis control : surveillance, planning, financing. WHO Library cataloguing-in-publication data.